缺血性疾病包括缺血性心脏病、脑缺血性疾病和外周动脉疾病等,是局部组织器官的血流受阻或受限导致组织缺血缺氧而发生的疾病,缺血缺氧可诱导炎症反应、氧化应激、内皮功能障碍,导致细胞坏死、凋亡和组织纤维化。这类疾病具有高发病率和高死亡率,严重威胁公众健康。基于促血管生成理念的治疗方案是热门临床研究方向,然而目前的血管生成机制尚不明确,缺乏有效治疗靶点,阻碍了这一治疗方案的应用。

内皮-间质转化(EndMT)是内皮细胞(EC)的动态生物学过程,参与缺血性疾病的发展,特征为内皮细胞丧失内皮特性(如血管形成)而获得间充质特征(如迁移能力),在病理条件下,缺氧、炎症、氧化应激、高血糖和低剪切应力可触发EndMT。部分EndMT(partial EndMT)是一种中间表型阶段,其特征是细胞保留或部分丧失内皮特征、并获得间充质特征,这种特殊表型在血管新生过程中发挥关键作用。
复旦大学附属中山医院、心脏病全国重点实验室葛均波院士、李华研究员团队在The Innovation(IF 25.7)上发表了研究论文“GTF2H4 regulates partial EndMT via NF-κB activation through NCOA3 phosphorylation in ischemic diseases”,发现转录因子IIH亚基4(GTF2H4)可通过与切除修复交叉互补组3(ERCC3)协同作用,促进EC在缺氧微环境中发生部分EndMT;GTF2H4可在部分EndMT期间磷酸化核受体共激活因子3(NCOA3)的1330位丝氨酸,促进NCOA3和p65的相互作用,激活NF-κB/Snail信号轴;GTF2H4在体内可显著促进缺血性损伤后的部分EndMT和血管生成。在研究中,作者使用汉恒生物提供的慢病毒在HMEC-1细胞系中过表达或敲低GTF2H4、ERCC3基因,以及携带Tie内皮特异性启动子的腺相关病毒(AAV)在小鼠离体主动脉环中过表达GTF2H4、在小鼠下肢过表达或敲低GTF2H4基因。

接下来,我们一起看看本研究的主要结果。
1.缺血/缺氧诱发部分EndMT
首先,作者构建了小鼠心肌梗死模型,通过免疫荧光染色分析发现,心肌梗死后内皮标记物血小板-内皮细胞黏附分子(CD31)和间充质标记物平滑肌肌动蛋白α(α-SMA)双阳性细胞显著增加。体外实验中,人微血管内皮细胞(HMEC-1)缺氧处理后逐渐表达α-SMA并丢失内皮标记物钙黏蛋白-5(VE-Cad);qPCR和Western Blot结果进一步证实,缺氧处理后CD31和VE-Cad表达下调,α-SMA、纤连蛋白(Fibronectin)表达上调。而用于评估内皮特征的成管实验结果显示,缺氧3天的HMEC-1仍保持管形成能力。这些结果表明,缺血缺氧诱导内皮细胞发生部分EndMT。
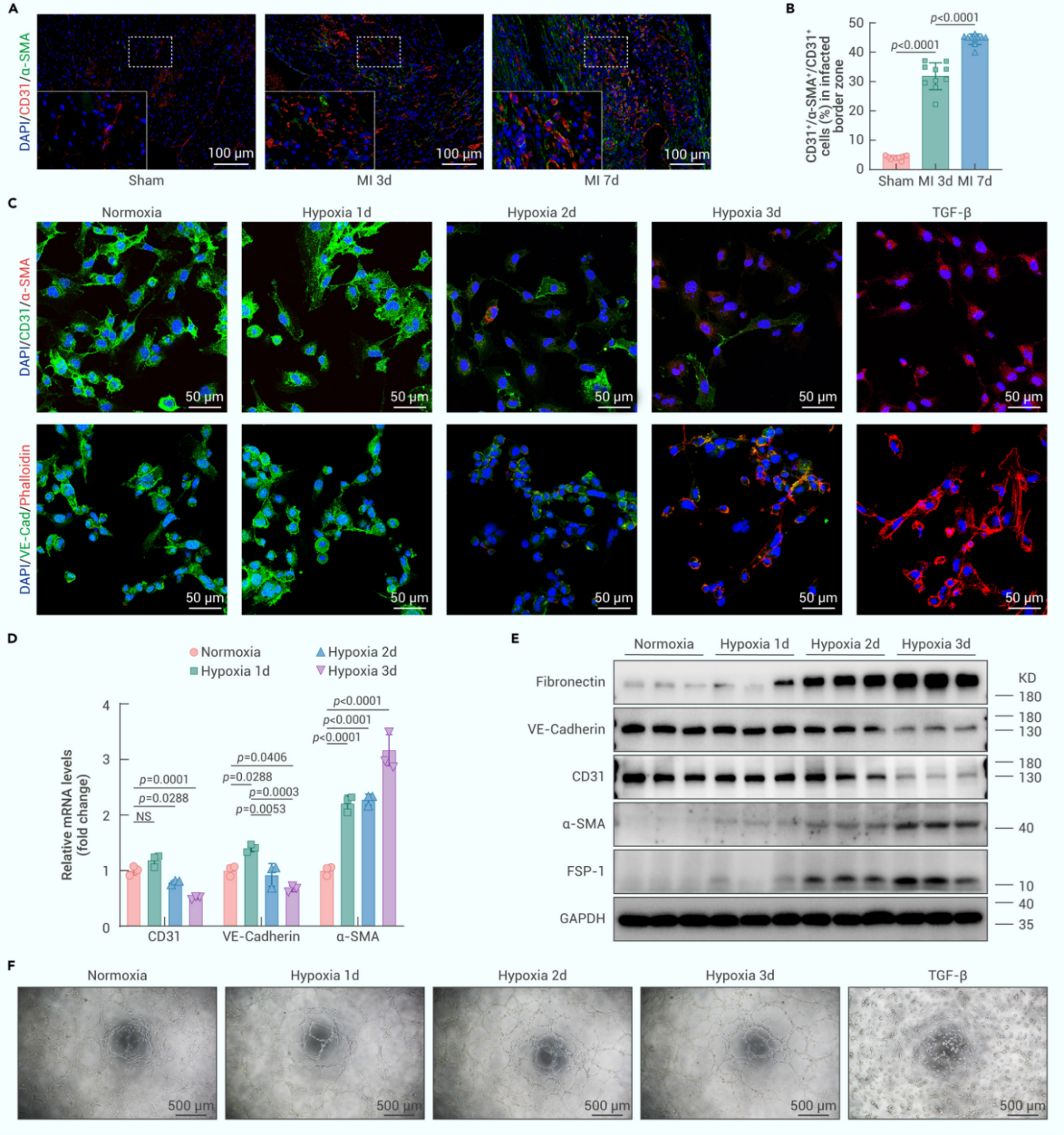
图1. 缺血/缺氧诱发部分EndMT
2. GTF2H4缓解缺氧诱导的微血管内皮细胞损伤,并促进缺氧诱导的部分EndMT
既往研究表明GTF2H4是缺血再灌注损伤早期阶段的活跃转录调节因子,于是,作者分析了心肌梗死患者经皮冠状动脉介入治疗(PCI)前后血液的转录组测序数据,并进一步结合GTEx数据库信息确认GTF2H4在内皮细胞中丰度较高,且因缺血/缺氧而下调。体外实验中,GTF2H4在缺氧诱导的HMEC-1和小鼠心脏微血管EC(MCMECs)中呈时间依赖性下调;CCK-8结果显示,GTF2H4过表达改善了HMEC-1细胞的缺氧损伤,而GTF2H4敲低加剧了损伤;Western Blot结果显示,GTF2H4过表达促进了缺氧条件下抗凋亡蛋白Bcl-XL的表达,并抑制促凋亡蛋白Bax和caspase3剪切体的表达,而GTF2H4敲低则相反。此外,GTF2H4过表达在缺氧条件下诱导α-SMA、Fibronectin、FSP-1(间充质标记物)的过表达,下调但保留了部分CD31和VE-Cad的表达,GTF2H4敲低则抑制了这些作用。这些结果表明GTF2H4缓解缺氧诱导的微血管内皮细胞损伤,并促进缺氧诱导的部分EndMT。
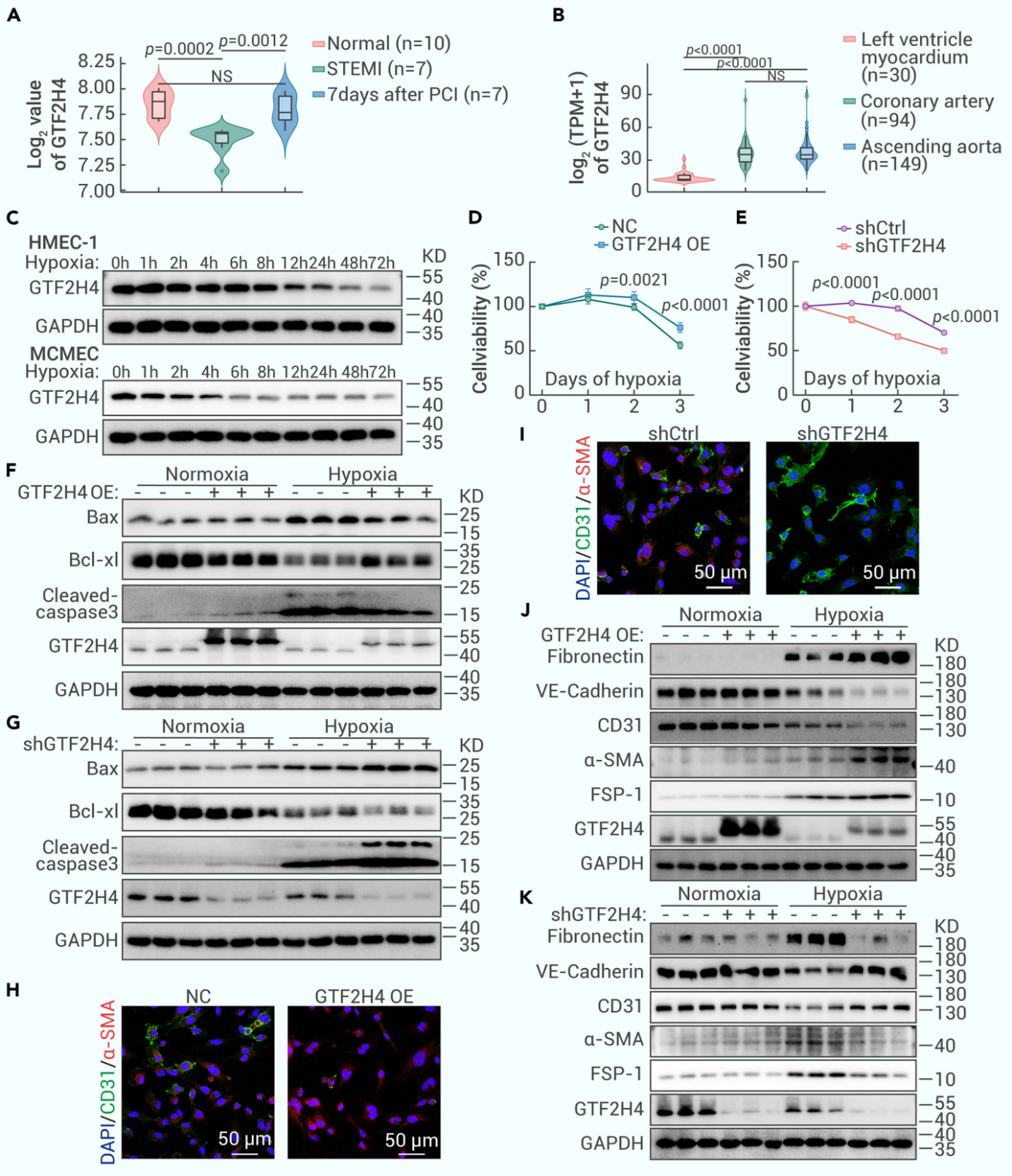
图2. GTF2H4缓解缺氧诱导的微血管内皮细胞损伤,并促进缺氧诱导的部分EndMT
3. GTF2H4增强部分EndMT期间的迁移并抑制管形成
为了研究GTF2H4在部分EndMT期间对细胞迁移和侵袭功能的作用,作者进行了划痕和Transwell实验,结果显示缺氧显着增强了HMEC-1细胞的迁移能力,GTF2H4的过表达在缺氧条件下进一步增加了细胞迁移,而GTF2H4敲低抑制了这一表型。主动脉环血管生成测定证明GTF2H4增强了缺氧诱导的血管芽的生成,此外,Western Blot结果显示GTF2H4可促进缺氧条件下促转移酶基质金属蛋白酶9(MMP-9)的蛋白表达。在管形成实验中,GTF2H4过表达轻微抑制了缺氧时的管形成,而GTF2H4敲低则促进该过程。上述实验表明GTF2H4通过驱动部分EndMT进程,增强细胞迁移/侵袭能力并导致细胞内皮特征部分丧失。
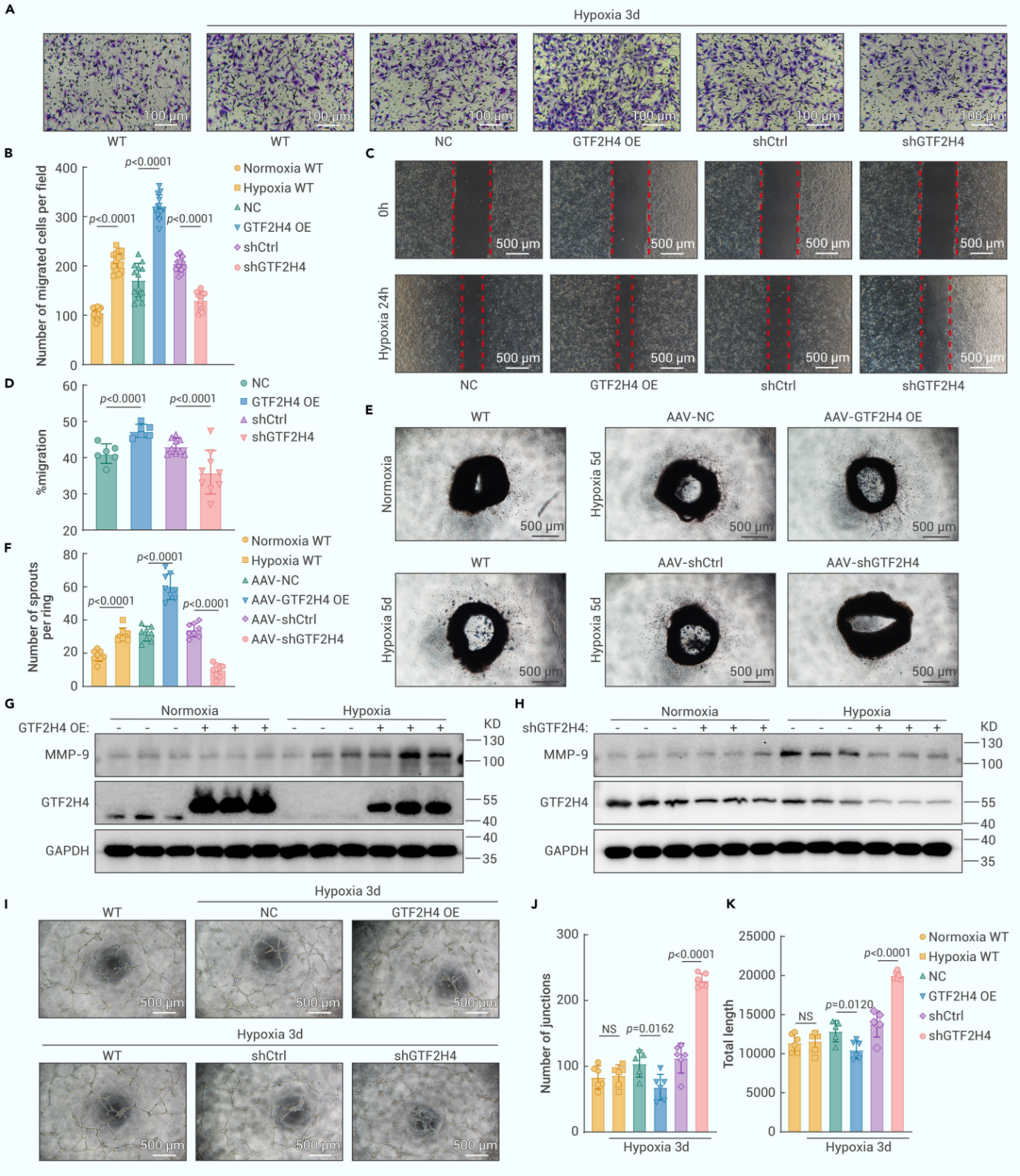
图3. GTF2H4增强部分EndMT期间的迁移并抑制管形成
4. GTF2H4在部分EndMT过程中通过自噬介导的降解调节ERCC3
接下来,作者在HMEC-1细胞中过表达或敲低GTF2H4,以阐明GTF2H4对缺氧诱导调节部分EndMT的分子机制。对4D Label Free蛋白质组学分析结果的重叠差异表达蛋白(DEP)筛选后,作者鉴定获得了下游的正调节靶分子——核苷酸修复交叉互补基因3(ERCC3),GTF2H4与该基因同属转录因子IIH(TFIIH)复合物亚基,且具有其结合结构域。Co-IP结果显示GTF2H4与ERCC3存在直接相互作用,qRT-PCR和Western Blot结果表明GTF2H4只在蛋白质水平上正向调控ERCC3表达,这表明GTF2H4可能影响ERCC3的蛋白稳定性。作者进一步使用蛋白酶体抑制剂MG132和自噬抑制剂3-MA处理GTF2H4过表达或敲低的HMEC-1细胞,通过蛋白质合成抑制剂环己酰亚胺(CHX)追踪实验,发现3-MA消除了GTF2H4过表达引起的ERCC3蛋白积累,以及GTF2H4敲低引起的ERCC3蛋白降解,而MG132无此效应。这些结果表明GTF2H4通过自噬介导的蛋白降解来调节ERCC3蛋白稳定性。
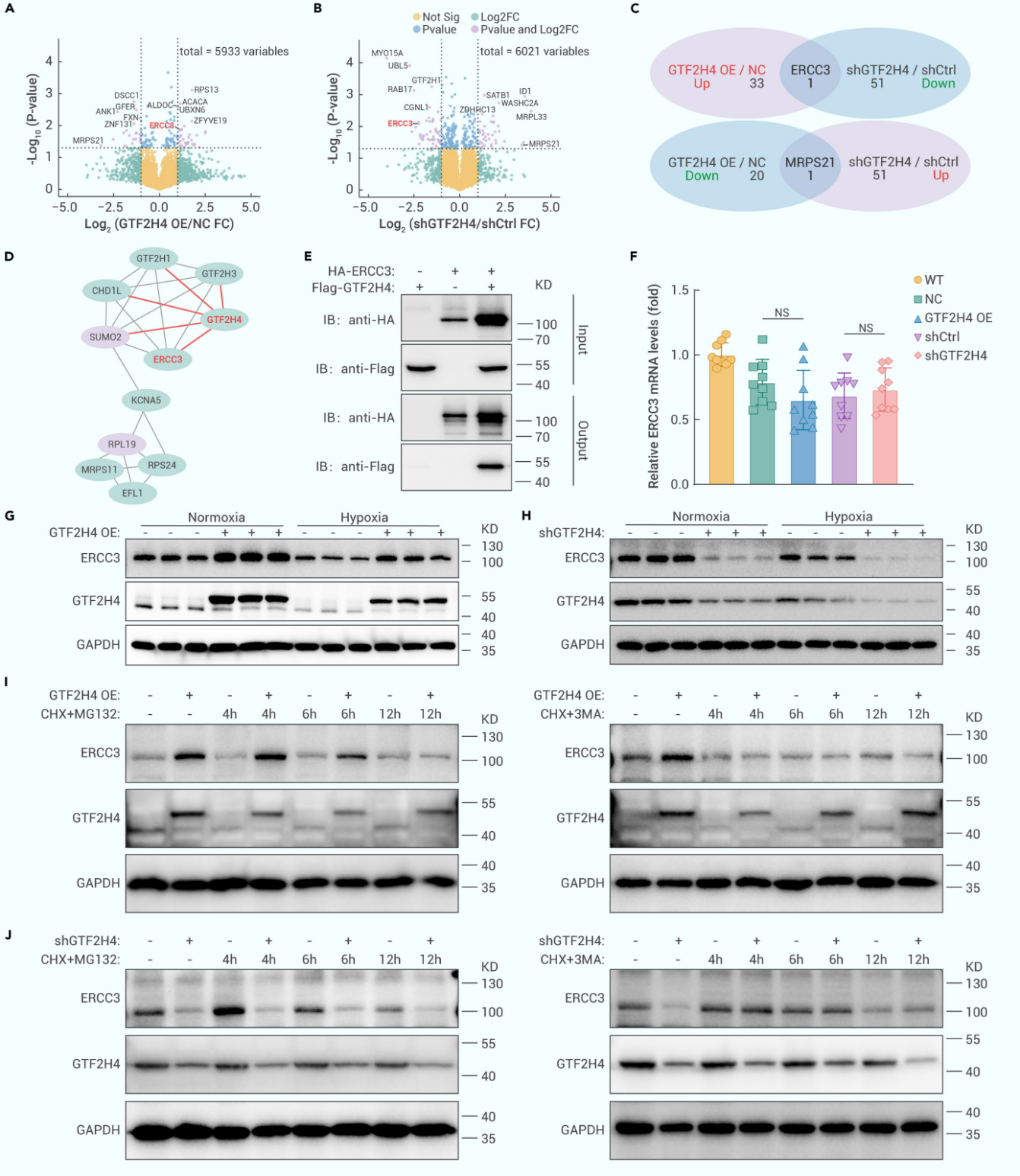
图4. GTF2H4在部分EndMT过程中通过自噬介导的降解调节ERCC3
5. 缺氧条件下GTF2H4介导的部分EndMT需要RCC3-GTF2H4E互作
为明确ERCC3在GTF2H4介导部分EndMT过程中的功能,作者在HMEC-1细胞中过表达或敲低了ERCC3基因。Western Blot结果显示ERCC3敲低抑制GTF2H4的表达,但ERCC3过表达不影响GTF2H4的表达,此外,ERCC3敲低抑制缺氧诱导的间充质标志物的表达并促进内皮标志物的表达;transwell和流式细胞术检测表明ERCC3敲低在缺氧条件下抑制细胞迁移,加剧细胞凋亡。而ERCC3敲低也逆转了缺氧条件下由GTF2H4过表达诱导的促迁移和抗凋亡作用,并逆转了GTF2H4过表达所引起的EndMT标志物表达变化。先前的研究证明GTF2H4的E310K/R314E突变可抑制其与ERCC3的互作,于是作者在HMEC-1细胞中过表达该GTF2H4突变体,与NC对照相比,GTF2H4 E310K/R314E突变与ERCC3的相互作用减少,GTF2H4突变体的过表达无法改变部分EndMT标志物表达水平。这些结果表明GTF2H4-ERCC3的相互作用是缺氧诱导的部分EndMT的必要条件。
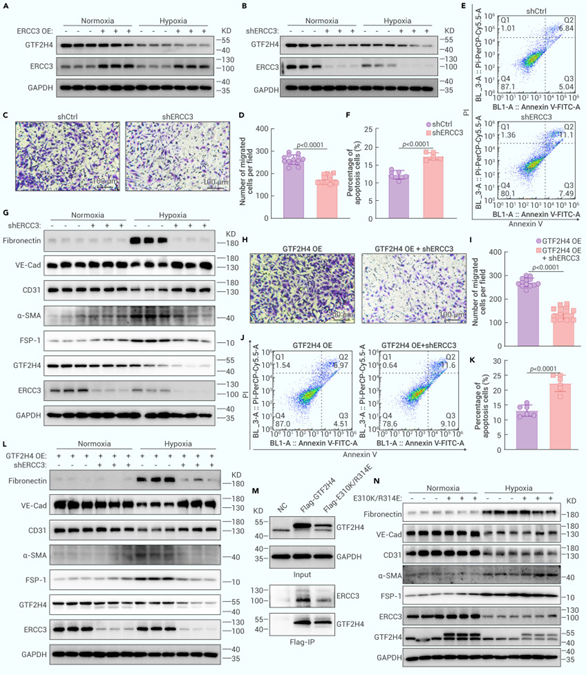
图5. 缺氧条件下GTF2H4介导的部分EndMT需要RCC3-GTF2H4E互作
6. GTF2H4通过NF-κB信号促进缺氧诱导的部分EndMT
为了研究GTF2H4调控部分EndMT的信号通路,作者通过磷酸化蛋白质组学分析了过表达或敲低GTF2H4的HMEC-1细胞,发现丝裂原活化蛋白激酶机制靶点(如Hippo、NF-κB和Notch)通路中存在磷酸化修饰差异。其中,作者重点关注了NF-κB通路,磷酸化抗体微阵列分析显示GTF2H4过表达增加了p65关键激活位点Ser536及其他NF-κB通路蛋白的磷酸化,降低了IκB激酶和IKK复合体的磷酸化。双荧光素酶报告基因检测表明GTF2H4过表达显著促进肿瘤坏死因子α(TNF-α)诱导的NF-κB启动子转录活性,而敲低则抑制了该活性。Western Blot结果显示在缺氧条件下,GTF2H4过表达显著上调P65的磷酸化和Snail的表达,GTF2H4敲低抑制了这些基因的表达,GTF2H4失活突变体也无法激活NF-κB信号,此外,NF-κB抑制剂Bay 11-7082逆转了GTF2H4过表达诱导激活的NF-κB信号。这些结果表明,GTF2H4通过NF-κB信号轴驱动缺氧诱导的部分EndMT。
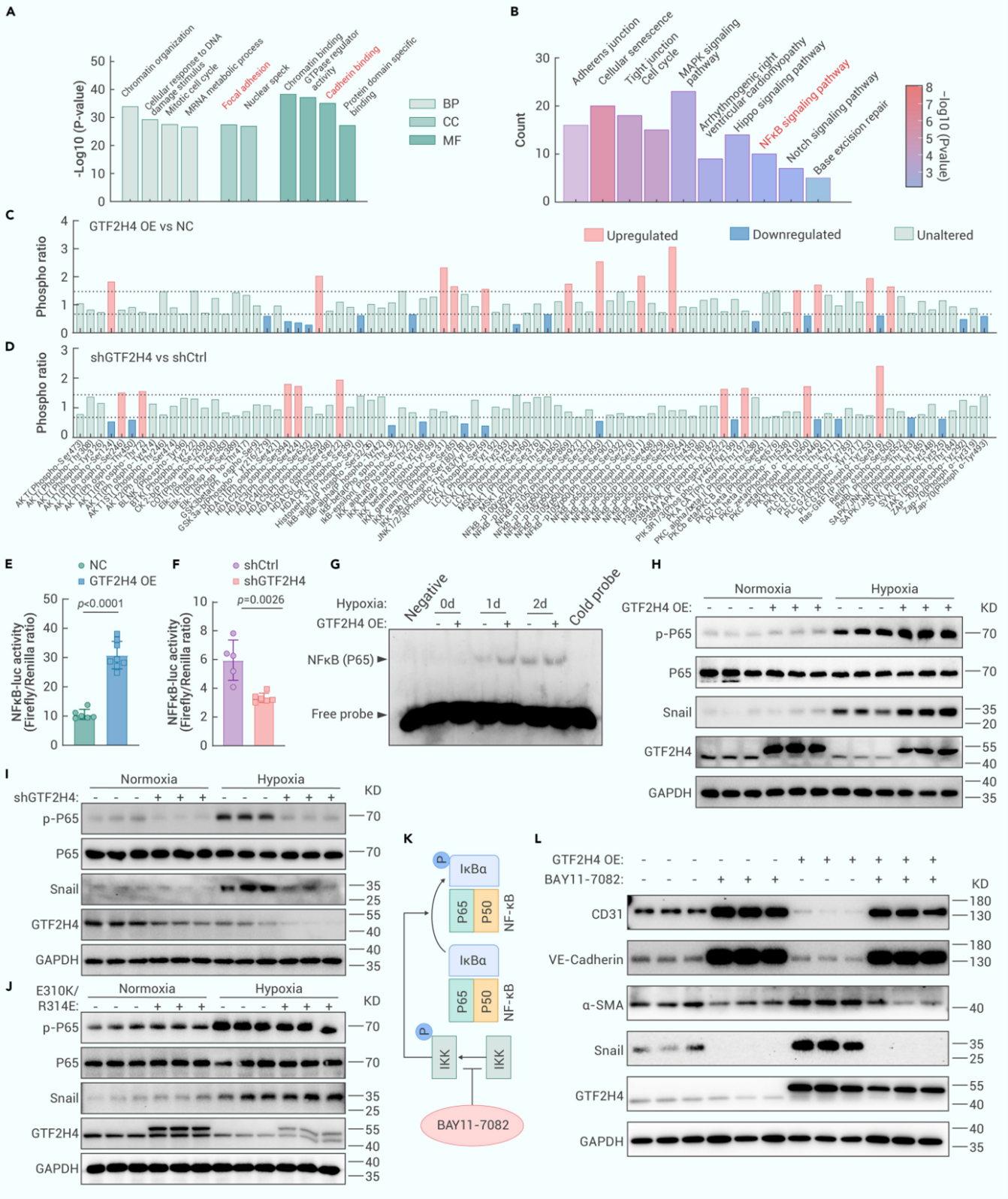
图6. GTF2H4通过NF-κB信号促进缺氧诱导的部分EndMT
7. GTF2H4通过NCOA3的S1330位磷酸化促进NF-κB通路激活
随后,作者分析了GTF2H4过表达或敲低实验中的DEP,发现NF-κB共核受体共激活因子3(NCOA3)的Ser1330磷酸化显著富集。在几项磷酸化突变实验中,作者发现NCOA3磷酸化缺陷突变(S1330A)诱导NF-κB启动子活性明显降低,NCAO3模拟磷酸化突变(S1330D、S1330E)可诱导与野生型相似的启动子活性。Co-IP实验进一步证实,S1330A突变抑制了COA3和NF-κB p65亚基之间的相互作用,而S1330E或S1330D突变的结合能力与野生型相似。此外,S1330A或S1330E突变均消除了GTF2H4过表达/敲低诱导的NF-κB启动子活性变化。这些结果表明NCOA3的S1330磷酸化对NF-κB的激活至关重要。
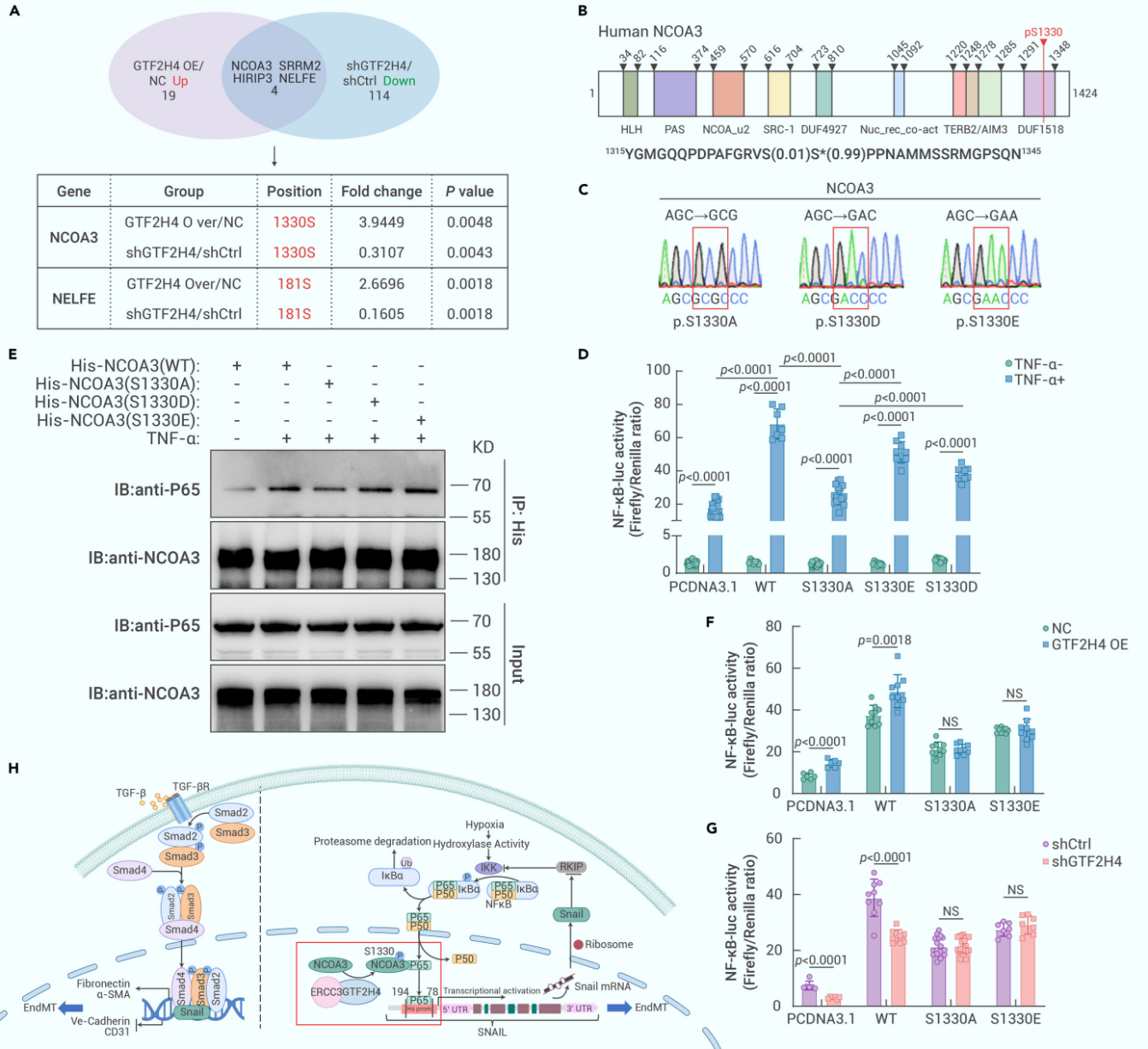
图7. GTF2H4通过NCOA3的S1330位磷酸化促进NF-κB通路激活
8. GTF2H4促进体内部分EndMT并改善缺血后损伤后血流恢复
最后,为了评估GTF2H4在体内对缺血损伤后恢复的影响,作者在下肢缺血小鼠模型的腓肠肌中特异性过表达或敲低了内皮细胞的GTF2H4。激光多普勒血流成像显示,与对照组相比,GTF2H4过表达组在后肢缺血7天内的肢体灌注恢复度增加,而GTF2H4敲低组肢体灌注恢复度减少。随后,作者通过免疫荧光染色、免疫组化染色和Western Blot分析发现,GTF2H4过表达可上调间充质标志物表达并激活NF-κB信号,而敲低则抑制这些变化。这些结果表明,GTF2H4在体内驱动部分EndMT进程,改善了缺血组织的血流重建能力。
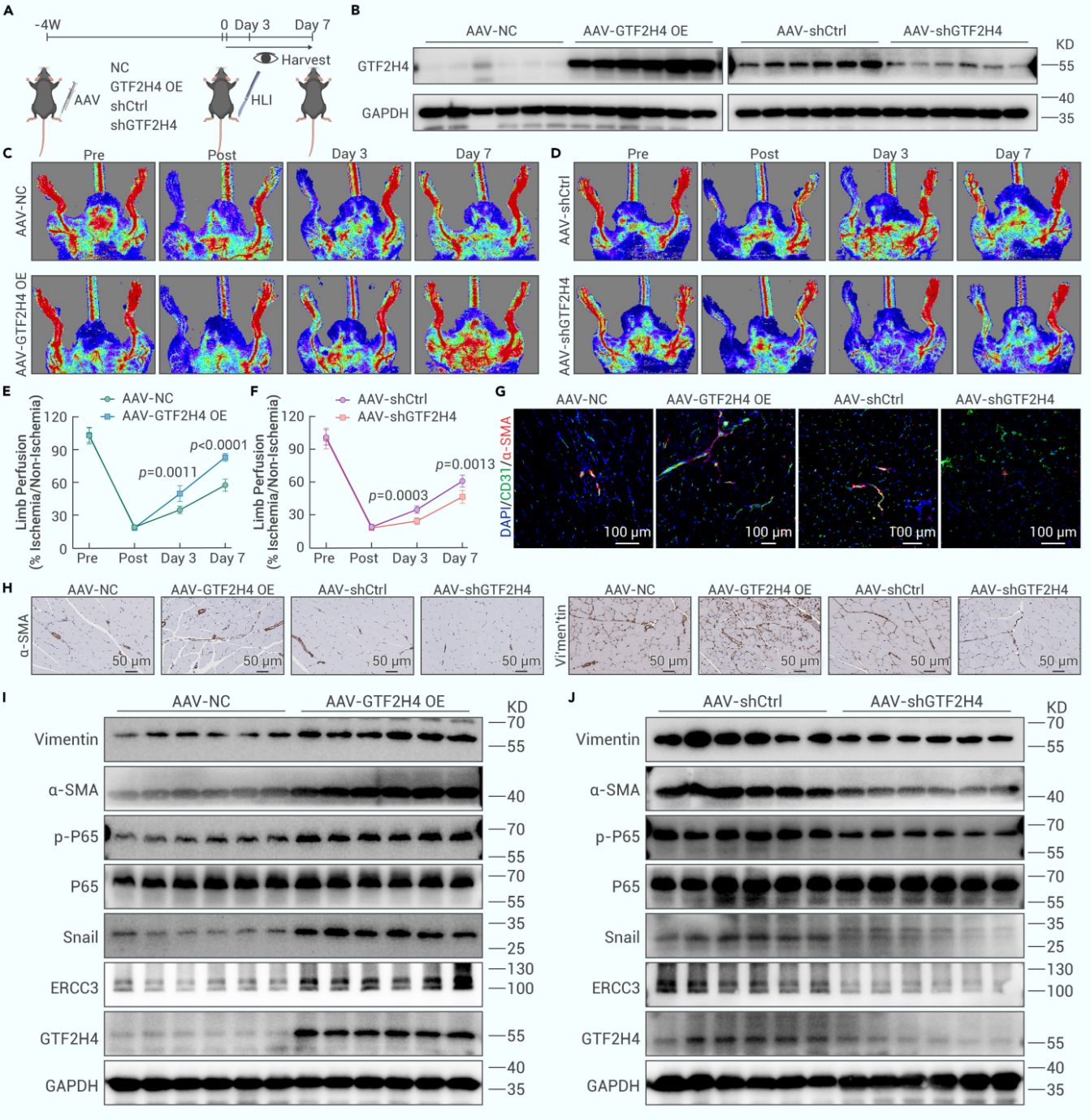
图8. GTF2H4促进体内部分EndMT并改善缺血后损伤后血流恢复
综上所述,本研究探索了GTF2H4的关键作用,发现GTF2H4与ERCC3形成功能复合物,通过激活NCOA3/NF-κB/Snail信号轴,介导了内皮细胞发生部分EndMT,这一机制在体外显着改善缺氧条件下的内皮细胞的活力,并增强迁移能力;在体内显著改善了缺血模型的血流灌注,促进新侧支血管新生。这一发现揭示了部分EndMT在缺血性疾病血管重建中的关键作用,为靶点治疗提供了新策略。









 8434
8434










