慢性胰腺炎(Chronic Pancreatitis,CP)是各种病因引起的胰腺组织和功能不可逆改变的慢性炎症性疾病,临床主要表现为反复发作的上腹部疼痛和胰腺内、外分泌功能不全,病理特征包括胰腺腺泡细胞破坏、间质纤维化等。在国内,CP发病率有逐年增高的趋势,人群中CP的发病率为每10万人9.62例,死亡率为万分之九。胰腺纤维化是胰腺炎的一个独特的组织病理学特征,表现为正常胰腺组织结构的丧失,并被细胞外基质(ECM)中丰富的纤维组织所取代,活化的胰腺星状细胞(PSCs)在CP的胰腺纤维化中起着至关重要的作用,但PSCs的激活机制尚不明确。
2023年8月25日,华中科技大学赵刚团队在Gastroenterology(IF=29.4)在线发表题为“Pancreatic acinar cells-derived sphingosine-1-phosphate contributes to fibrosis of chronic pancreatitis via inducing autophagy and activation of pancreatic stellate cells”的研究论文,研究结果表明受损胰腺腺泡细胞iPACs(injured pancreatic acinar cells)中活化的SPHK1(sphingosine kinase 1)/S1P(sphingosine-1-phosphate)通路通过调控S1PR2/AMPK/mTOR通路诱导PSCs自噬和活化,促进CP的纤维化发生,并且缺氧微环境可能参与了PACs与PSCs在CP发病中的作用。值得注意的是,本研究使用了汉恒生物提供的自噬双标慢病毒。
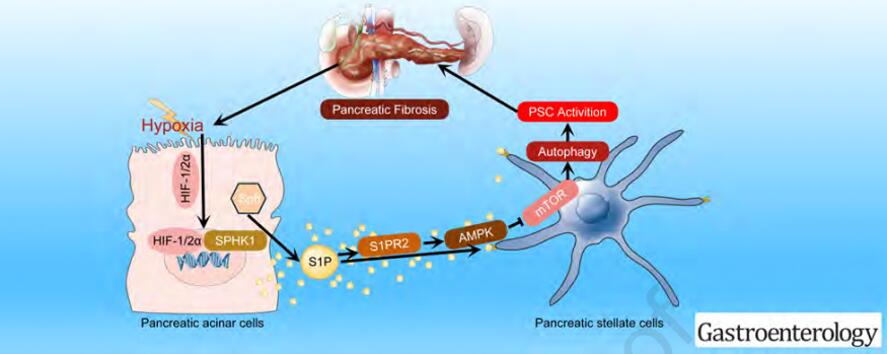
图1 调控机制模型图
首先,作者通过腹腔注射cerulein或胰管结扎(PDL)构建了两种小鼠模型,各项实验指标都表明模型小鼠表现出了与CP一样的慢性炎症、纤维化、PSC激活等表型,说明cerulein或PDL可以引起CP,CP小鼠模型构建成功。S1P是SPH在SPHK1作用下的磷酸化产物,在CP小鼠观察到SPHK1和S1P表达量显著增加,并且SPHK1主要是在腺泡细胞区域表达,在PSC几乎不表达。推测SPHK1/S1P信号轴在CP发展中起作用。所以SPHK1-/-和SPHK1-KD小鼠用cerulein或PDL诱导CP后检测相关指标,发现SPHK1敲除或敲低后,S1P表达量下调,纤维化相关基因、炎症相关基因的表达量也显著下调,胰腺的萎缩和损伤都得到了缓解,说明SPHK1/S1P信号轴可能促进CP的纤维化形成。
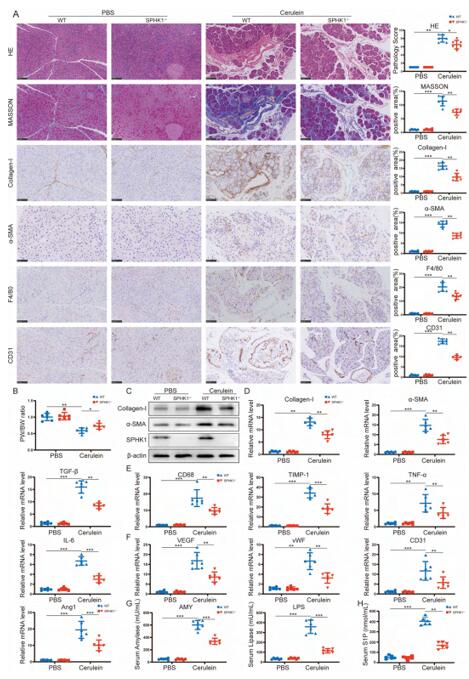
图2 SPHK1-/-和SPHK1-KD小鼠CP发病机制
PAC和PSC之间的相互作用对于胰腺纤维化的进展至关重要,对离体细胞PAC和内皮细胞、巨噬细胞分别进行CCK(cholecystokinin)刺激,模拟CP中的细胞损伤,只有PAC在CCK刺激后SPHK1表达上调,培养基中S1P含量也增加,并且能诱导PSC激活,排除了在动物体中内皮细胞、巨噬细胞激活PSC的可能。作者进一步收集了各种处理条件下的iPAC培养基(CM)来处理PSC,发现被CCK刺激过的WT小鼠PAC的CM即WT-CMCCK促进PSC中纤维化相关蛋白α-SMA高表达以及增强了PSC的迁移能力,而SPHK1敲除小鼠的PAC(SPHK1-/--CMCCK)、SPHK1敲低的PAC(siSPHK1- CMCCK)、SPHK1抑制剂PF-543处理的PAC经CCK刺激后的CM对PSC没有相似的激活作用,CCK直接刺激PSC,PSC的激活和迁移能力也没有显著改变,但是如果S1P重组蛋白直接添加到PSC的培养基中,PSC可以被激活,甚至可以逆转SPHK1-/--CMCCK、siSPHK1-CMCCK和PF-543-CMCCK对PSC激活的抑制作用。这说明PSC的激活是由iPAC中的SPHK1和S1P驱动的。
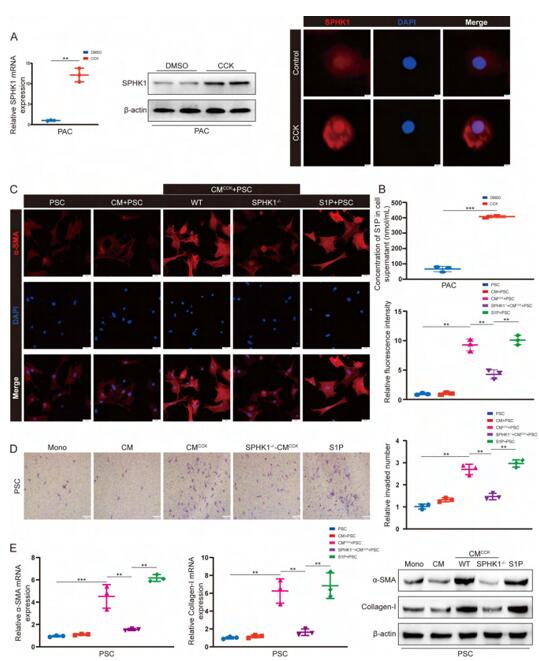
图3 CCK损伤PACs通过SPHK1/S1P信号诱导PSCs激活
那么是哪种受体接受iPAC释放的S1P信号激活PSC呢?实验数据表明,CP小鼠PSC的S1PR2表达量显著上调,而且S1P处理后的PSC中只有S1PR2表达显著增加,推测S1PR2是S1P诱导PSC激活的受体。随后,作者验证了这一推测,S1P或CMCCK诱导的PSC激活和迁移能力增加会被siS1PR2或S1PR2抑制剂JTE013所抑制,S1PR2的确是PSC特异性S1P受体,对于诱导PSC激活至关重要。
S1P或CMCCK诱导的PSC中还观察到LC3B-II与LC3B-I的比值明显增大,Beclin1和ATG5的表达增加,SQSTM1的表达减少,即PSC自噬增强,通过自噬抑制剂CQ(CQ能阻断自噬小体与溶酶体融合)预处理可显著逆转SQSTM1的减少,但不能降低Beclin1和ATG5点表达,这种自噬和活性表型的改变与自噬诱导剂雷帕霉素(RAP)作用结果相似。利用汉恒生物的LC3自噬双标慢病毒LV-mRFP-GFP-LC3检测发现S1P或CMCCK诱导的PSC的红色斑点增加,但绿色斑点没有增加,和RAP诱导的自噬荧光结果一样,而经自噬抑制剂CQ预处理后,RAP和S1P或CMCCK诱导的PSC红色、绿色以及merge后的黄色斑点均显著增加,siS1PR2、siATG5和JTE013能明显抑制S1P或CMCCK诱导的自噬和自噬体的形成,说明S1P通过S1PR2以RAP的方式诱导PSCs自噬内流,CQ能阻断这一过程。细胞免疫荧光和Transwell测定结果显示抑制自噬的同时也抑制了S1P或CMCCK诱导的PSC的激活和迁移,说明S1P诱导PSC自噬对PSC的活化有重要作用。
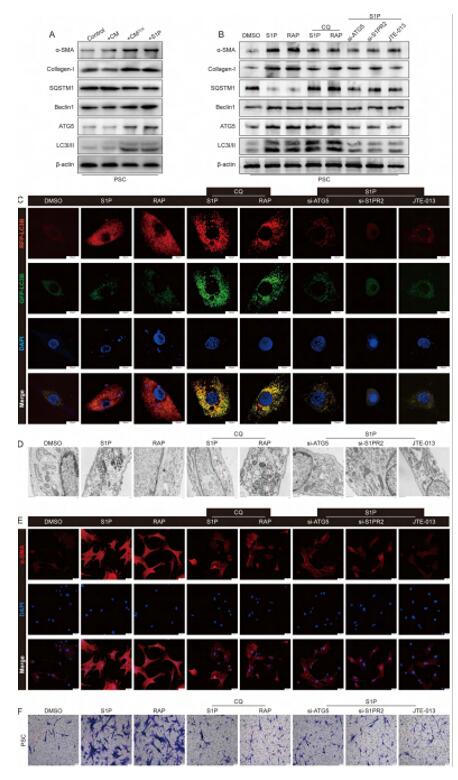
图4 S1P通过S1PR2介导PSCs自噬激活
既然已经证明S1P在调节PSCs自噬流中的作用与RAP相似,那么合理猜测S1P也是通过mTOR途径发挥作用的,数据显示S1P或CMCCK诱导的PSC中p-mTOR和p-S6的表达显著降低,mTOR上游调节因子AMPK的磷酸化水平显著增加,JTE-013或AMPK抑制剂可以逆转激活的AMPK/mTOR通路,结合前面的实验结果,说明S1P/S1PR2信号通路通过调节AMPK/mTOR通路促进PSCs的自噬。
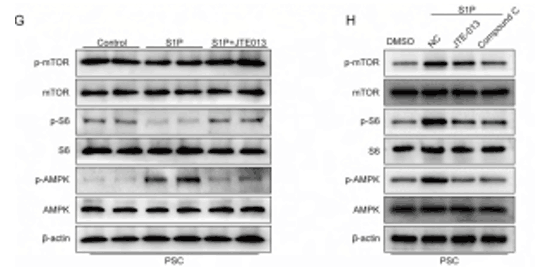
图5 S1P/S1PR2调节AMPK/mTOR通路
胰腺纤维化形成后,胰腺微环境处于缺血缺氧状态。根据报道,缺氧环境中,HIF-1α或HIF-2α可诱导SPHK1表达,作者发现CP小鼠组织的HIF-1α和HIF-2α表达上调,缺氧处理的PAC中SPHK1和S1P表达也显著增加,随后实验证实了HIF-1α和HIF-2α可以结合SPHK1启动子,并且缺氧条件会增加HIF-1α和HIF-2α与SPHK1启动子的结合来促进SPHK1的转录,进而PAC CMhypoxia激活了PSC,siHIF-1α、siHIF-2α的PAC经缺氧处理后的CM刺激PSC,细胞的激活和迁移都受到了抑制。
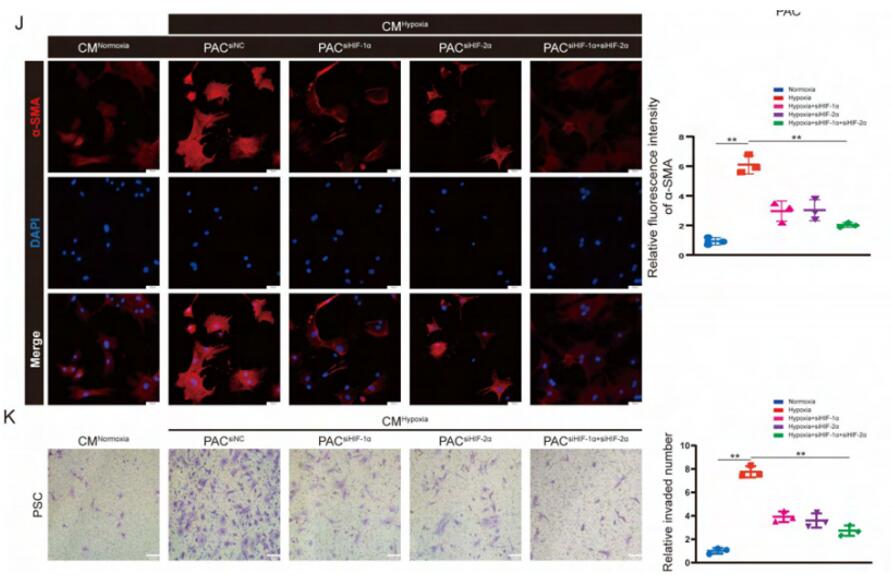
图6 缺氧诱导的HIF-1α和HIF-2α促进了SPHK1转录以及PSC的激活
PAC和PSC的体外实验初步证明了SPHK1/S1P/S1PR2信号传导在PSC的激活中发挥作用。在动物体内,PF-543和JTE-013治疗均能显著抑制CP小鼠胰腺纤维化、血管生成和炎症相关基因的表达,改善胰腺外分泌功能,缓解CP小鼠的发病,说明抑制SPHK1/S1P/S1PR2信号传导可显著减轻CP小鼠的胰腺纤维化。
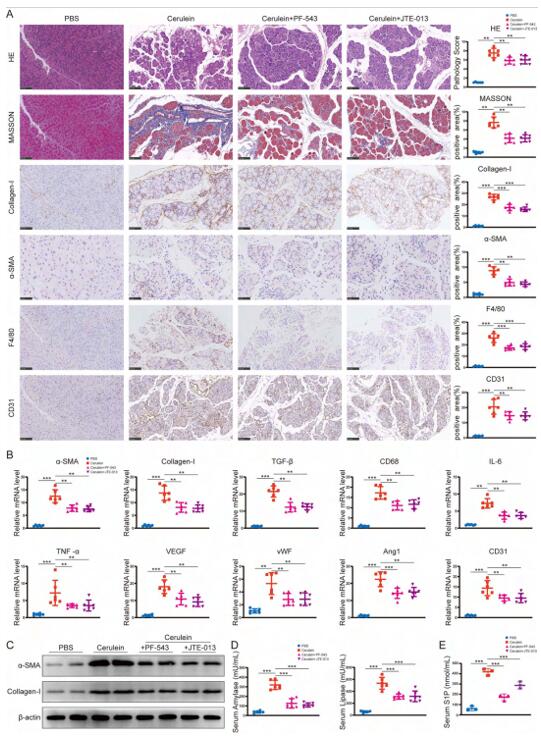
图7 抑制SPHK1/S1P/S1PR2信号通路可减轻CP小鼠的发病机制和纤维化
综上所述,PACs中SPHK1催化SPH生成S1P,S1P通过S1PR2释放并活化PSCs。活化的PSCs分泌大量ECM,促进胰腺纤维化和局部缺氧,从而加重PACs损伤。SPHK1/S1P通路为未来的治疗提供潜在的靶点,SPHK1和S1PR2抑制剂在本实验的应用也为临床治疗CP提供新思路。









 8325
8325










