上一期干货为大家详细介绍了DNA的甲基化修饰,本期我们继续为大家介绍组蛋白的甲基化修饰。

什么是组蛋白?
组蛋白是存在于真核生物细胞核中的一类碱性蛋白质,是染色质的主要成分。组蛋白分为核心组蛋白(H2A、H2B、H3和H4)和接头组蛋白(H1和H5)两大类。每种核心组蛋白各有两个分子,共同形成八聚体结构,构成核小体核心,缠绕约146bp的DNA。连接组蛋白位于核小体之间的连接DNA区域,功能是稳定核小体结构,促进染色质高级折叠(如30nm纤维)。它们不仅是DNA的“包装材料”,还通过化学修饰动态调控基因表达,参与细胞分裂、DNA修复等关键生物学过程。
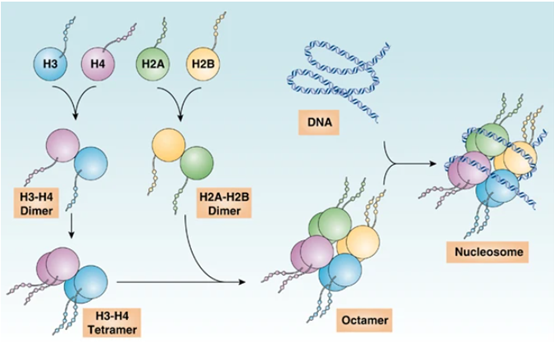
图1、组蛋白结构和核小体组装[1]
组蛋白甲基化
组蛋白甲基化是指在组蛋白H3、H4的特定赖氨酸或精氨酸残基上添加甲基基团的化学修饰过程。与组蛋白磷酸化和乙酰化等修饰不同,甲基化修饰不改变其电荷,而是作为称为“组蛋白读取器”的特定结合蛋白的停靠位点。组蛋白甲基化修饰,如同DNA序列之外的“暗物质”,在生命活动中扮演着至关重要的角色。虽然它不改变DNA序列本身,却能调控基因的表达,影响细胞命运,甚至决定个体发育和疾病发生。
甲基化修饰包括甲基化与去甲基化两个层面:甲基化即在氨基酸侧链上添加甲基,去甲基化即祛除侧链上添加的甲基。根据发生甲基化的氨基酸残基不同,可分为赖氨酸甲基化修饰和精氨酸甲基化修饰。
组蛋白赖氨酸甲基化的修饰
赖氨酸是含有一个ε-氨基的碱性氨基酸,其甲基化修饰主要发生在组蛋白H3和H4的特定赖氨酸残基上。组蛋白赖氨酸上的甲基化存在三个不同的水平:单甲基化、二甲基化和三甲基化。这种修饰在从单细胞生物到哺乳动物的不同物种间高度保守,并且与基因激活或抑制有关,具体作用取决于靶位点。例如组蛋白H3第4位、36位、第79位赖氨酸(H3K4、H3K36、H3K79)的甲基化与基因激活相关,而发生在H3K9、H3K27、H4K20的甲基化修饰则与基因的沉默相关。
组蛋白内赖氨酸残基的甲基化受甲基转移酶(KMT)和去甲基化酶(KDM)的严格调控,以维持细胞命运和基因组稳定性。早在1960年代,RNA合成被证明受组蛋白中赖氨酸ε-氨基甲基化的调节[2],然而直到2000年,ThomasJenuwein及其同事才取得了具有里程碑意义的发现,确定了第一个催化组蛋白甲基化的甲基转移酶(KMT):人和小鼠SUV39H1(KMT1A)。在发现KMT1A后,通过与酶促SET结构域的同源搜索鉴定了数十个KMT。SET结构域是一个130个氨基酸的催化结构域,最初发现在Su(var)3-9、E(z)(zeste增强子)和trithorax中是保守的。另一类不含SET的KMT仅有KMT4一个成员,虽然它们的催化结构域不同,但是都使用S-腺苷-L-蛋氨酸(SAM)作为甲基供体。KMT对底物内赖氨酸选择和甲基化程度具有高度酶特异性,例如,KMT1A/B催化组蛋白3赖氨酸9从单甲基化状态(H3K9me1)到三甲基化状态(H3K9me3),而KMT1C则具有双甲基化偏好性,将其从单甲基化状态(H3K9me1)催化成双甲基化状态(H3K9me2)。KMT2A(也称为MLL1)可催化H3K4甲基化为H3K4me2,但当其与内源性相互作用蛋白结合时,KMT2A也可以催化H3K4为H3K4me3,因此,KMT可以是高度特异性的,但它们的相互作蛋白可以改变其靶赖氨酸或活性程度。
关于去甲基化酶的争论持续多年,直到本世纪初,YangShi及其同事确定了第一个组蛋白KDM:LSD1/KDM1A[3]。KDM1A与其他类似的辅阻遏物复合物相关,表明该蛋白质是候选阻遏物,包含一个黄素腺嘌呤二核苷酸(FAD)依赖性胺氧化酶结构域,该结构域可去甲基化H3K4me2和H3K4me1并调节基因表达。发现KDM1A之后,又发现了另一类KDM,该酶类利用JmjC结构域通过甲基氧化催化去甲基化。JmjC蛋白依赖于α-酮戊二酸、分子氧和Fe(II)作为去甲基化的辅助因子。KDM2A/B、JHDM1A和JHDM1B是最早记录的JmjC去甲基化酶,它们和KDM3A-KDM3C是通过测定色谱级分从甲基化组蛋白中释放甲醛的能力来鉴定的[4]。KDM2和KDM3不能移除赖氨酸的三甲基化修饰,KDM4A-KDM4D是第一个被发现的能移除三甲基化修饰的去甲基化酶,他们可以使移除H3K9me3/H3K9me2、H3K36me3/H3K36me2和H1.4K26me3/H1.4K26me2的甲基化修饰,却不能移除H3K9me1或H3K36me1的甲基化修饰。
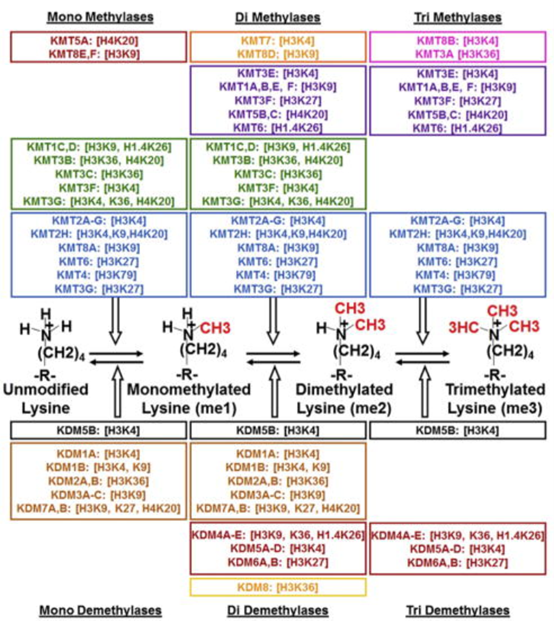
图2、调控赖氨酸甲基化修饰及调控特性[5]
组蛋白精氨酸甲基化修饰
精氨酸的甲基化修饰发生在胍基氮中,主要可以催化三种不同方式的甲基化:ω-NG-单甲基精氨酸(MMA);ω-NG,N G-不对称二甲基精氨酸(ADMA);和ω-NG,N′G 对称二甲基精氨酸(SDMA)。当这些甲基添加到精氨酸残基中时,它们都不会改变其正电荷,但它们可能通过消除潜在氢键的形成和改变精氨酸侧链的体积来影响蛋白质-蛋白质相互作用。组蛋白精氨酸甲基化调节许多不同的细胞过程,包括细胞信号传导、转录调控、RNA代谢和DNA修复。
调控组蛋白精氨酸甲基化修饰的酶主要为蛋白质精氨酸甲基转移酶(ProteinArginineMethyltransferases,PRMTs)。根据甲基化产物的不同,PRMTs分为I型、II型和III型[6]。I型PRMTs:催化形成单甲基精氨酸(MMA)和不对称二甲基精氨酸(ADMA),主要成员为PRMT1、PRMT3、PRMT4和PRMT6,催化H3R2和H4R3上ADMA形成,导致转录激活和核糖体生物合成,PRMT1是人细胞中的主要甲基转移酶,可修饰~90%的甲基化精氨酸残基。PRMT1通常识别富含甘氨酸-精氨酸的区域内的精氨酸,该基序存在于许多RNA或DNA结合蛋白中 。II型PRMT(PRMT5)和(PRMT7)会导致H3R8和H4R3上形成SDMA,从而导致转录抑制。III型仅有一个成员,催化形成单甲基化(MMA)。
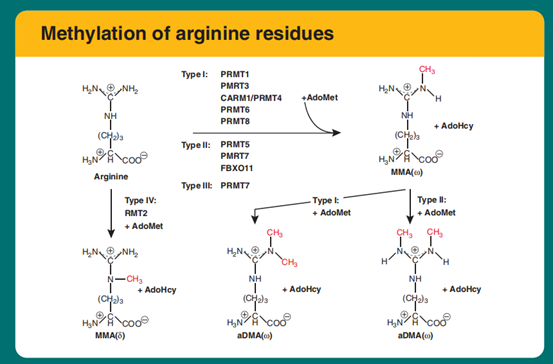
图3、精氨酸甲基化修饰[6]
到目前为止,已鉴定PAD4和JMJD6两种组蛋白精氨酸去甲基化酶。PAD4被认为是一种去甲基化酶,PAD4靶向组蛋白H3和H4中的多个位点,包括被共激活因子组蛋白-精氨酸甲基转移酶CARM1(H3R17)和PRMT1(H4R3)甲基化的位点。去甲基亚胺化不是“真正的”去甲基化反应,它会去除甲基化,但不会逆转甲基化。产生中性瓜氨酸,与未甲基化的精氨酸相比,其化学性质有很大不同,这个过程不足以维持精氨酸循环。此外,由于PAD4也能够催化非甲基化精氨酸,因此PAD4是否是一种严格的组蛋白去甲基化酶存在争议 。JMJD6通过去除甲基直接将甲基精氨酸转化为精氨酸。JMJD6以前被描述为巨噬细胞和树突状细胞质膜的磷脂酰丝氨酸受体,能够通过去除甲基来去甲基化H3R2me2和H4R3me2。另外,据报道含有加氧酶的Fe(II)和2OG依赖性JmjC的一个亚群,即组蛋白赖氨酸去甲基化酶,也能够催化精氨酸去甲基化。另外,KDM3A、KDM4E、KDM5C和KDM6B也表现出去甲基化催化活性,KDM4E和KDM5C则被证明可以催化在H3R2、H3R8、H3R26(KDM4E)和H4R3位点甲基化的组蛋白肽的去甲基化。
组蛋白甲基化修饰在各种疾病领域的研究
组蛋白甲基化动力学在许多生物过程中起着重要作用,包括细胞周期调节、DNA损伤和应激反应、发育和分化,生物体中的组蛋白在甲基化/去甲基化之间维持着微妙的平衡,这种平衡被打破时,可能导致许多疾病的发生。
H3K4me3是研究最多的组蛋白甲基化修饰,在癌症中经常发现H3K4甲基化转移酶基因的突变[7]。涉及编码KMT2A的基因染色体重排,例如易位、倒位或基因融合,导致 N 端KMT2A与其他蛋白质融合,是急性白血病的常见原因,KMT2A的这些重排通常通过募集转录延伸复合物、破坏正常的造血分化并导致白血病的发展而导致基因表达失调。尽管融合蛋白的KMT2A部分失去了催化活性,但KMT2A-AF9融合蛋白的存在会导致H3K4me2和H3K4me3水平异常。编码KMT2B的基因突变也存在于几种类型的癌症中,例如淋巴瘤、乳腺癌、结直肠癌和其他恶性肿瘤。编码KMT2B和KMT2C的基因的失活突变是癌症中编码SET1/MLL亚基的最常见突变基因(两者在大约10%的所有类型的癌症中都发生突变)。尽管编码SETD1A、SETD1B和MLL2的基因在癌症中不经常发生突变,但这些基因的突变与各种类型的癌症有关,包括白血病、乳腺癌和肺癌 。SETD1A和SETD1B的失调则会影响基因表达程序,并以H3K4me3依赖性或不依赖性方式促进肿瘤的发生和发展。
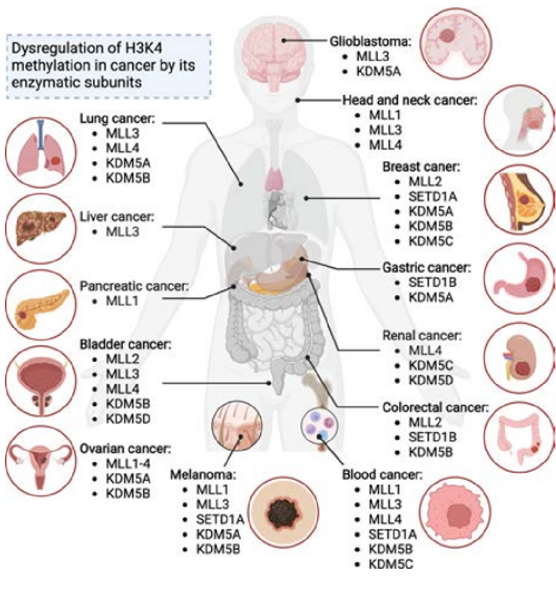
图4、各种癌症中参与调控H3K4甲基化修饰的酶[7]
KDM5A-D与各种类型的癌症有关。这些去甲基化酶的失调可导致H3K4me3模式异常和基因表达谱改变,从而导致癌症的发生、进展和对治疗的耐药性 。研究表明,KDM5组蛋白去甲基化酶亚家族成员KDM5A是导致多种人类疾病(尤其是癌症)的原因。KDM5A可以消除H3的第四个赖氨酸中的二甲基和三甲基部分(H3K4me2/3),从而导致转录的激活或抑制 。此外,由NUP98和KDM5A重排产生的融合基因NUP98-KDM5A介导造血细胞增殖,并通过去甲基化H3K4me2/3改变髓红细胞生成分化。
LSD1也在许多癌症中异常表达,阻碍癌细胞分化,促进癌细胞增殖、转移和侵袭,并与预后不良有关。在造血和淋巴肿瘤中,包括急性髓性白血病(AML)、急性淋巴细胞白血病、T细胞非霍奇金淋巴瘤和霍奇金淋巴瘤,都发现了LSD1的过表达。LSD1还与多种实体瘤有关,包括非小细胞肺癌、神经母细胞瘤、胰腺癌、前列腺癌和乳腺癌,抑制LSD1的作用可能会减少或阻止这些肿瘤的细胞生长[8]。
包括甲基化在内的多种组蛋白修饰物已被证实智力障碍综合征有关[9]。例如,在 77%的Sotos综合征患者中发现了组蛋白甲基转移酶NSD1的截断体;而H3K9甲基转移酶EHMT1突变则引起9q亚端粒缺失综合征(9qSTDS),EHMT1的单倍体不足被认为会导致智力障碍。在 X连锁智力低下(XLMR)中鉴定到至少7种潜在的甲基修饰酶或甲基结合蛋白,分别是MECP2(一种甲基CpG结合蛋白)、JARID1C/SMCX95(一种H3K4去甲基化酶)、PHF8(一种H3K9/H4K20去甲基化酶)、BCOR(一种锚蛋白重复序列,含有与多梳组蛋白复合)、ATRX(一种H3K9me3结合蛋白)、PHF6(一种含有PHD的蛋白)和BRWD3(一种含有bromo和WD重复序列的蛋白)。MECP2失衡会导致Rett综合征和神经元染色质结构的整体变化,这些变化伴随着组蛋白甲基化模式的整体变化。ATRX与H3K9me3结合,并在将变体组蛋白与端粒结合中发挥作用。针对一名严重智力障碍患者的研究中,发现该患者同时携带MeCP2和ATRX的重复,这表明这两种蛋白质在调节认知方面可能存在功能联系。
组蛋白甲基化和甲基修饰蛋白在调节生物体寿命和组织衰老中发挥重要作用。成体干细胞中稳定和动态甲基标记之间的平衡丧失可能导致个体组织功能下降。组蛋白甲基化水平随年龄变化的证据增加了整体获得或丢失稳定甲基标志物可能导致生物体衰老的可能性。例如,大鼠肝脏中H4K20me3水平随年龄增长而增加 ,秀丽隐杆线虫体细胞中H3K27me3水平随年龄增长而降低。异染色质在老年人的细胞或患有过早衰老疾病Hutchinson-Gilford早衰综合症(HGPS)的患者细胞中减少,而HGPS患者个体来源的细胞显示失活的X染色体上H3K27me3降低,H3K27三甲基转移酶EZH2[10]的水平降低。这些结果表明,异染色质的整体减少,以及随之而来的许多在年轻健康个体中本应沉默的甲基化修饰调控基因的失调和错误表达,可能在衰老中起因果作用。
组蛋白甲基化和去甲基化与动脉粥样硬化发生和发展密切相关[11]。动脉粥样硬化斑块中的H3K4甲基化增加,而VSMC和炎症细胞中H3K9和H3K27的甲基化降低。在颈动脉粥样硬化病变内的巨噬细胞中,lnc_000048通过减弱LSD1活性来促进MAP2K2转录,从而导致H3K4me2的积累,并最终诱导下游炎症因子。抑制KDM5活性会增加H3K4me3并显着减少心血管疾病患者血管组织中EC的增殖、迁移和管形成。组蛋白甲基转移酶EZH2在动脉粥样硬化斑块组织中升高,并通过介导H3K27me3沉默基因表达。敲低GAS5可能促进胆固醇的反向转运,并通过减少EZH2介导的对ATP结合盒转运蛋白A1(ABCA1)的转录抑制来最终阻止动脉粥样硬化的进展。此外,EZH2还调节DNA甲基转移酶1的表达,随后促进 Abca1 启动子的DNA甲基化,导致 Abca1 基因沉默。在动脉粥样硬化的发展过程中,DOT1L表达上调,它诱导的H3K79me2可以增加胞质CCL5和CXCL10的表达,从而促进VSMC的表型转换。
组蛋白的甲基化研究远不止于此,据pubmed数据显示,自上世纪60年代首次报道组蛋白甲基化以来,已有超过3万篇相关文献发表,近15年来年均发表数量超过1500篇。汉恒生物专营病毒包装十余载,建立了庞大的基因研究现货工具库,可为广大研究人员提供组蛋白甲基化修饰相关基因的过表达病毒现货。若有其他病毒包装需求或疑问,也欢迎随时咨询汉恒生物微信公众号或拨打官网技术服务热线:400-092-0065。
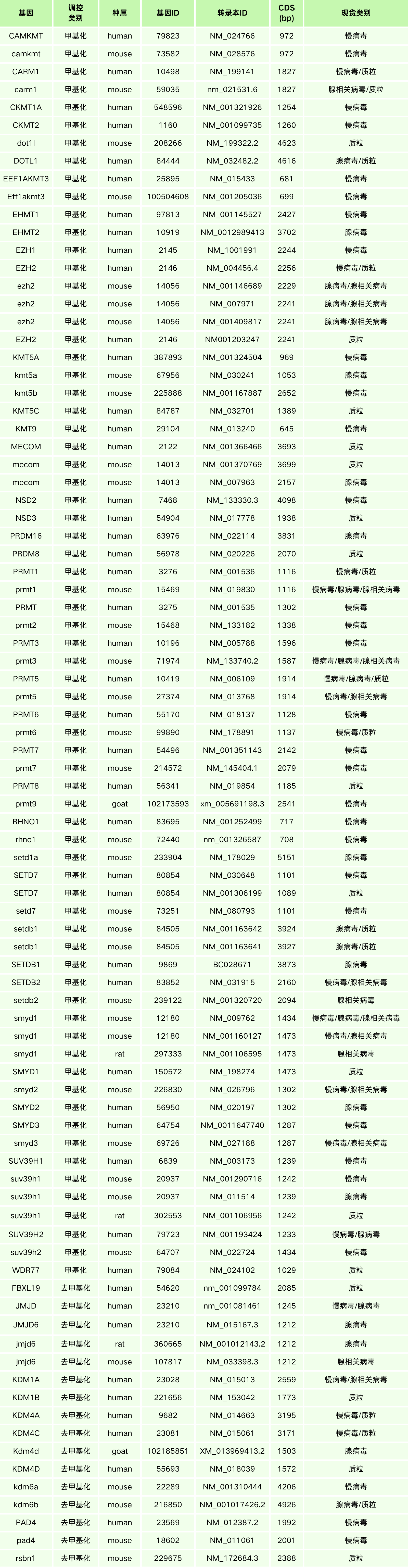
汉恒生物组蛋白甲基化相关产品现货
参考文献
1.Chen, R., Kang, R., Fan, XG. et al. Release and activity of histone in diseases. Cell Death Dis 5, e1370 (2014). https://doi.org/10.1038/cddis.2014.337
2.Allfrey, V.G., and Mirsky, A.E. (1964). Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science 144, 559.
3.Shi, Y., Sawada, J., Sui, G., Affar, B., Whetstine, J.R., Lan, F., Ogawa, H., Luke, M.P., Nakatani, Y., and Shi, Y. (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738.)
4.Tsukada, Y., Fang, J.,Erdjument-Bromage, H., Warren, M.E., Borchers, C.H., Tempst, P., and Zhang, Y. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816)
5.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell (2012) 48(4):491–507. doi: 10.1016/j.molcel.2012.11.006 ]
6.Katz, J. E., Dlakic, M. and Clarke, S. (2003). Automated identification of putative methyltransferases from genomic open reading frames. Mol. Cell. Proteomics 2, 525-540.
7.Wang H., Helin K., 2024. Roles of H3K4 methylation in biology and disease. Trends Cell Biol. 0. 10.1016/j.tcb.2024.06.001
8.Wan, W., Peng, K., et al. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α [J]. Oncogene 36, 3868–3877 (2017).
9.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–57. doi: 10.1038/nrg3173
10.Shumaker DK, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8. doi: 10.1073/pnas.0602569103.
11.Ji Y, Chen Z, Cai J. Roles and mechanisms of histone methylation in vascular aging and related diseases. Clin Epigenetics. 2025 Feb 23;17(1):35. doi: 10.1186/s13148-025-01842-y. PMID: 39988699; PMCID: PMC11849368









 8434
8434










