2025年5月21日,北京大学张岩/肖瑞平研究团队在《Circulation》(IF=35.6)上发表了题为“Intracellular L-PGDS-derived 15d-PGJ2 inhibits CaMKII through lipoxidation to alleviate cardiac ischemia/reperfusion injury”的研究论文。该研究揭示了心肌缺血/再灌注(ischemia/reperfusion,I/R)损伤过程中心肌细胞死亡和心脏损伤的新机制,为心血管疾病的机制研究与临床干预提供了新的思路。值得注意的是,在本研究中,作者使用汉恒生物提供的AAV9-cTNT-L-PGDS腺相关病毒,成功在小鼠心脏中特异性过表达L-PGDS。

缺血性心脏病是全球范围内发病率和死亡率最高的疾病之一,对公共健康构成重大威胁。再灌注治疗是目前心肌梗死的标准临床策略,然而该治疗方式可能诱发缺血/再灌注(I/R)损伤,导致额外的心肌细胞死亡和不可逆的心肌损伤,最终进展为心力衰竭。花生四烯酸(Arachidonic acid, AA)是一种广泛存在于体内的多不饱和脂肪酸。已有研究表明,AA及其代谢产物在炎症、氧化应激和细胞死亡等多种生理与病理过程中发挥关键作用,并与包括心血管疾病在内的多种疾病密切相关。然而,AA代谢酶在心脏I/R损伤中的具体作用机制尚未完全阐明。
在该研究中,作者通过分析心肌梗死后的小鼠心脏组织及细胞模型,发现脂质运载蛋白型前列腺素D2合成酶(L-PGDS)及其产物15-二聚体前列腺素J2(15d-PGJ2)在I/R损伤后显著降低,而这一变化与心脏功能障碍和细胞死亡密切相关。进一步机制研究表明,15d-PGJ2通过与Ca/钙调蛋白依赖性激酶II (Ca2+/calmodulin-dependent protein kinase II, CaMKII)结合并诱导其C495位点发生脂氧化修饰,从而抑制其活性和磷酸化水平,进而减轻I/R损伤引起的心肌细胞死亡和心脏损伤。此外,研究还发现, L-PGDS过表达或外源性给予15d-PGJ2治疗能够有效改善心脏功能,减少细胞死亡,并减轻DNA损伤。
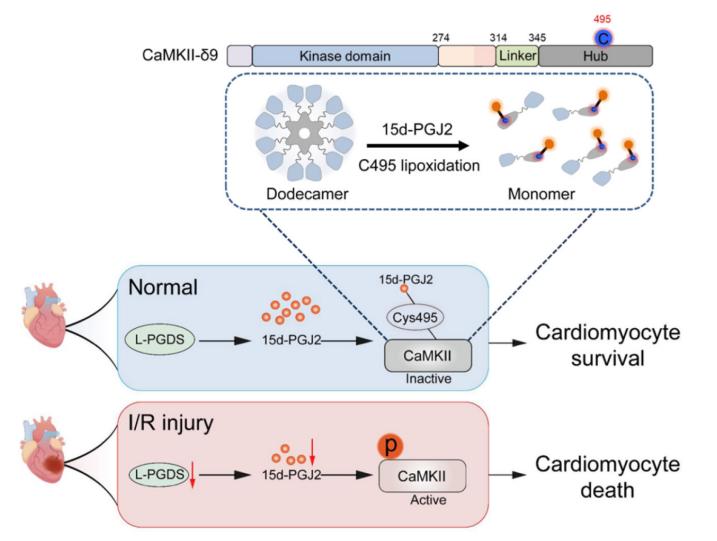
图1. L-PGDS/15d-PGJ2/CaMKII信号通路在I/R损伤中的保护机制图
下面,我们一起来了解具体的研究内容:
研究成果
1.L-PGDS 在I/R损伤中表达下调
作者通过分析小鼠I/R损伤模型心脏组织的转录组测序数据,对AA代谢通路相关基因的表达谱进行筛选。结果显示,编码L-PGDS的基因Ptgds在I/R损伤4 h和24 h的心脏组织中显著下调。随后,作者在体内和体外多个细胞模型中验证这一结果:L-PGDS在I/R损伤后不同灌注时长(4 h, 24 h,2 w)的小鼠心脏组织中的表达均下降;同时,在原代新生大鼠心室肌细胞(NRVMs)、成年大鼠心肌细胞(ARVMs)、人胚胎干细胞来源的心肌细胞(ES-HCMs)进行缺氧/复氧(hypoxia/reoxygenation, H/R)处理(模拟体内I/R损伤)后,L-PGDS的表达亦均下调。这些结果表明,I/R损伤诱导的L-PGDS下调主要源于心肌细胞的变化。
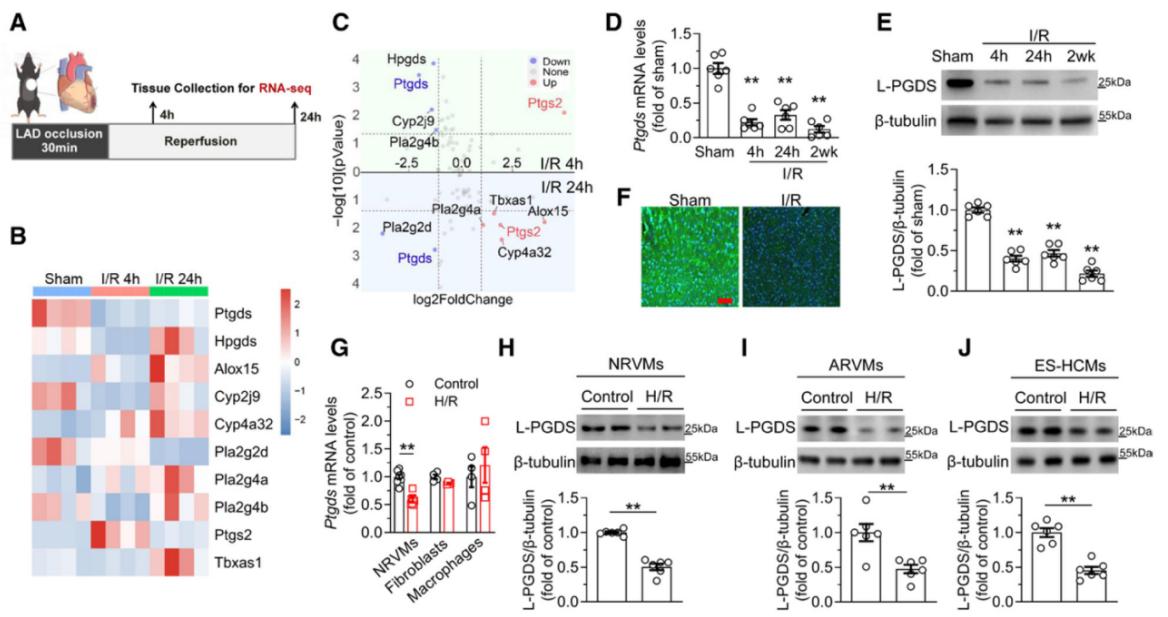
图2. L-PGDS在I/R损伤心脏中表达下调
2. L-PGDS是心肌细胞抵抗H/R诱导细胞死亡所必需的关键因子
为明确L-PGDS在I/R损伤中的功能,作者构建了Ad-L-PGDS腺病毒,在NRVMs、ARVMs和ES-HCMs中过表达L-PGDS后进行H/R处理。结果显示,在这些细胞中过表达L-PGDS可显著降低细胞损伤标志物-乳酸脱氢酶(LDH)的水平,并抑制细胞凋亡关键指标Caspase 3/7的活性,提示L-PGDS对心肌细胞具有明显的保护作用;相反,敲低L-PGDS则增强细胞对H/R的敏感性,加剧损伤程度。这些结果表明,L-PGDS是心肌细胞抵抗H/R诱导细胞死亡的关键保护因子。
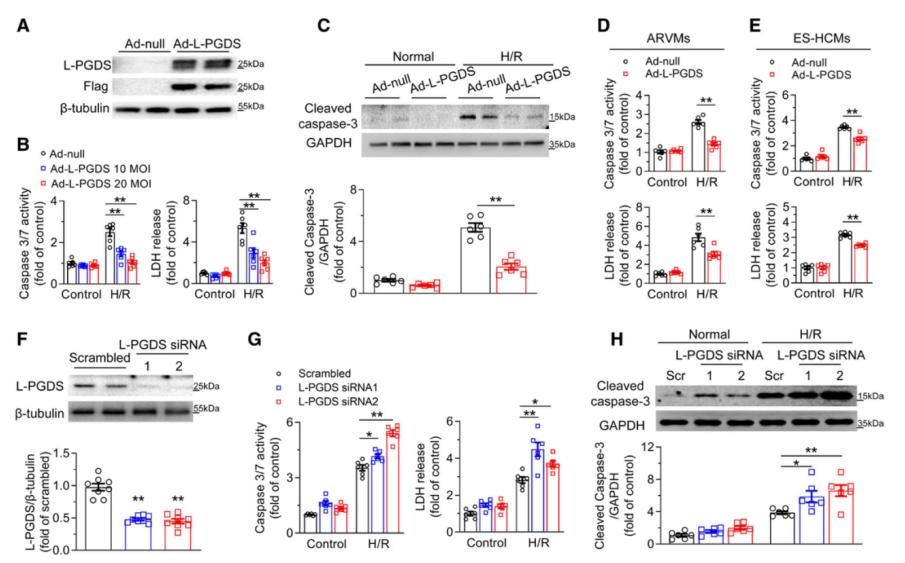
图3. L-PGDS是心肌细胞抵抗H/R诱导细胞死亡所必需的关键因子
3. 上调L-PGDS可改善I/R诱导的心脏损伤、心肌重构和心力衰竭
为了在体内验证L-PGDS在I/R损伤中的功能,作者利用腺相关病毒9型(AAV9)结合心脏特异性肌钙蛋白T (cTNT)启动子,构建了心肌细胞特异性过表达L-PGDS的转导系统(AAV9-cTNT-L-PGDS)。在正常生理状态下,AAV-L-PGDS注射组与AAV-null对照组相比,心脏形态、心功能及心肌细胞存活率均无显著差异,表明L-PGDS过表达在体内具有良好的安全性。在小鼠急性心脏I/R损伤模型(30 min缺血/24 h再灌注)中,L-PGDS过表达显著减小心肌梗死面积,减少心肌细胞凋亡和DNA损伤,有效缓解了心肌细胞死亡。在慢性心脏I/R损伤模型(30 min缺血/4 w再灌注)中,L-PGDS过表达同样改善心脏收缩功能(如左室射血分数),减轻心肌纤维化,抑制心室重构。上述结果表明,心肌细胞中L-PGDS的上调在心脏I/R损伤中发挥核心保护作用。
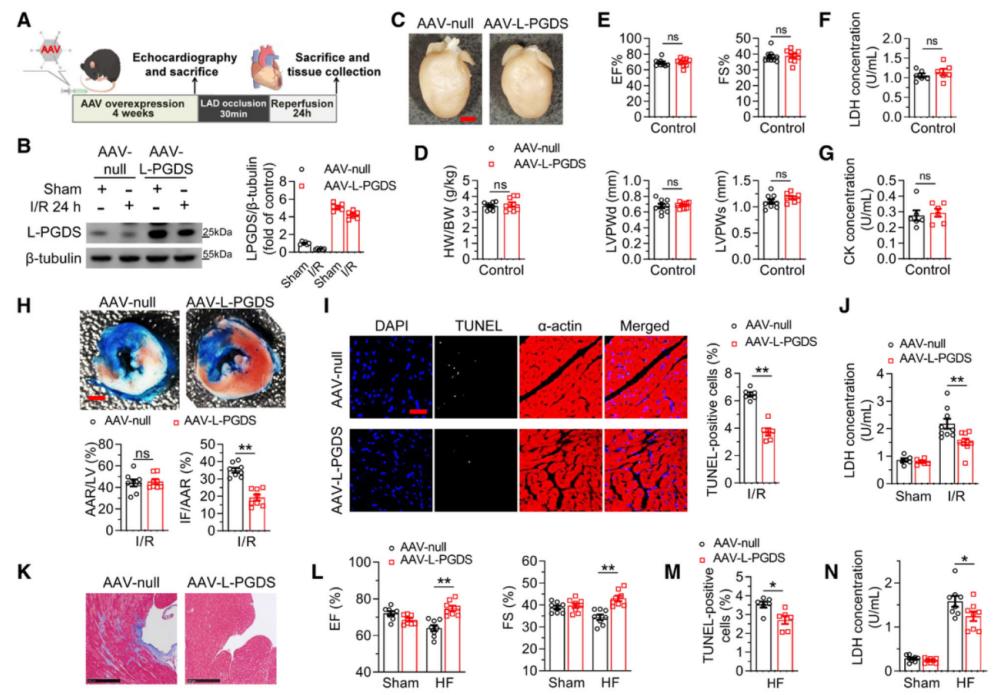
图4. L-PGDS上调减轻I/R导致的心肌损伤、心脏重构及心衰
4. I/R通过降低15d-PGJ2水平诱发心肌损伤
为了探究L-PGDS介导的心脏保护机制,作者进一步研究了其代谢产物在I/R损伤中的作用。前列腺素合成起始于磷脂酶A2(PLA2)从细胞膜磷脂中AA,随后AA经环氧合酶(COX)催化生成PGH2。L-PGDS作为COX下游的关键酶,进一步将PGH2转化为PGD2,后者通过自发脱水反应依次生成PGJ2和稳定的15d-PGJ2。利用液相色谱-串联质谱(LC-MS/MS)对I/R小鼠血清中COX通路的主代谢物分析,在4 h和24 h再灌注后,只有15d-PGJ2水平显著降低,这与心脏组织中L-PGDS的变化趋势一致。ELISA检测进一步证实,I/R损伤后小鼠缺血心肌组织和血清中的15d-PGJ2水平均下降。此外,在心肌梗死患者PCI术(经皮冠状动脉介入治疗)后,血浆中15d-PGJ2水平也显著下降。体外实验表明,补充15d-PGJ2可显著减轻H/R诱导的NRVMs、ARVMs和ES-HCMs损伤。体内实验中,再灌注前给予低剂量15d-PGJ2也有效减少了心肌梗死面积、细胞凋亡和DNA损伤。综上,I/R损伤通过下调L-PGDS减少15d-PGJ2生成,削弱其对心肌的保护作用,最终导致心肌细胞死亡;补充15d-PGJ2可逆转这一过程,为临床干预提供了新策略。
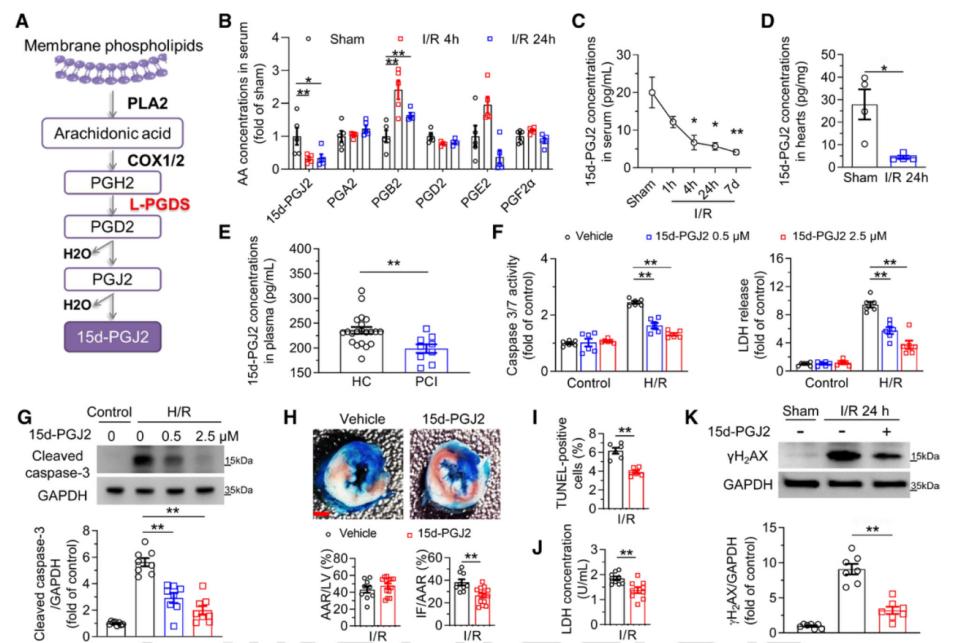
图5. I/R损伤显著降低15d-PGJ2水平
5.15d-PGJ2是内源性CaMKII抑制剂
接着,作者利用生物素标记的15d-PGJ2进行蛋白质组学分析,发现CaMKII是15d-PGJ2的直接作用靶点。功能实验显示,15d-PGJ2可以剂量依赖方式抑制CaMKII-δ9的激酶活性,且对其他亚型(α、β、γ)选择性较低。并且,给予15d-PGJ2可显著减少CaMKII活化及其介导的心肌细胞死亡。总之,这些结果表明15d-PGJ2作为内源性抑制剂直接与CaMKII结合。
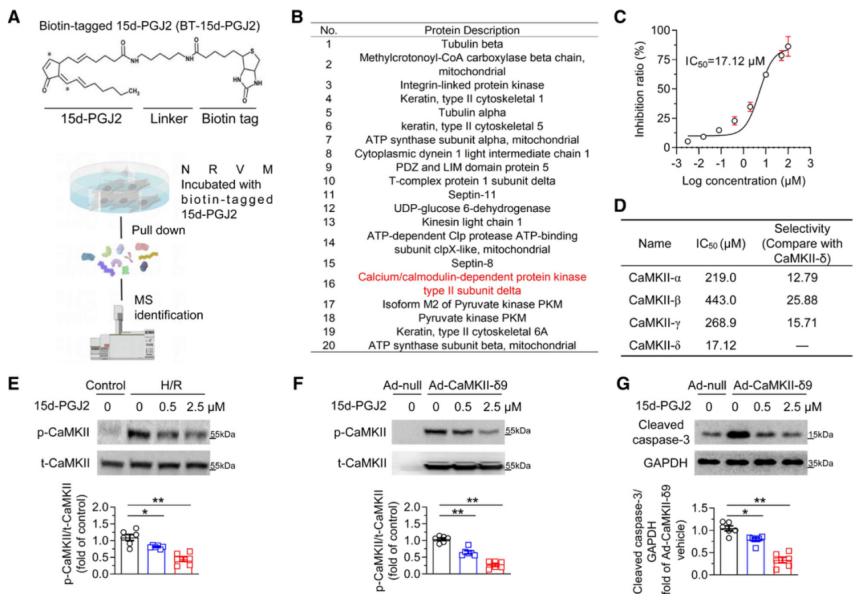
图6. 15d-PGJ2是内源性CaMKII抑制剂
6.L-PGDS/15d-PGJ2信号轴抑制I/R诱导的CaMKII激活
接下来,作者研究了L-PGDS/15d-PGJ2信号轴对I/R损伤后CaMKII磷酸化的调控作用。结果显示,15d-PGJ2显著抑制I/R损伤诱导的CaMKII的磷酸化。且这一作用不依赖于其已知受体(PPARγ或DP2),提示15d-PGJ2可能通过直接机制发挥功能。体内与体外实验均显示,L-PGDS过表达能够提高15d-PGJ2水平,并显著降低CaMKII的磷酸化水平;相反,敲低L-PGDS则加重CaMKII的磷酸化。这些结果表明,15d-PGJ2通过不依赖受体的方式抑制CaMKII磷酸化,进而发挥心脏保护作用。
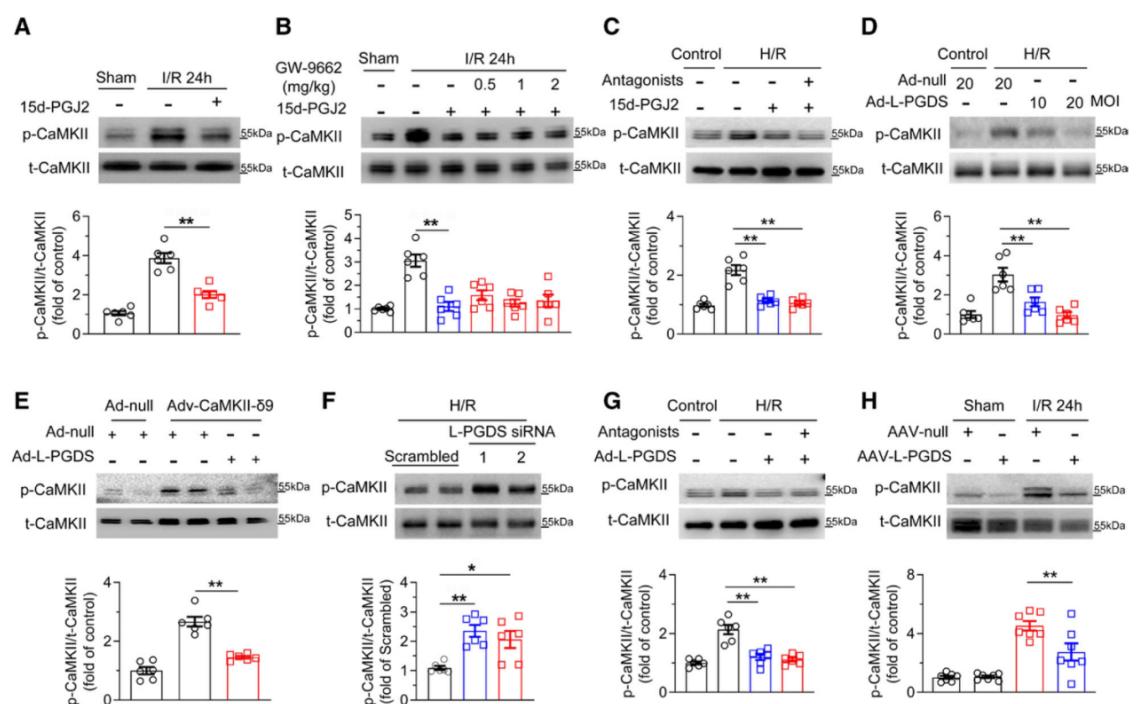
图7. L-PGDS/15d-PGJ2信号轴抑制I/R诱导的CaMKII激活
7.15d-PGJ2通过诱导CaMKII-δ9 C495位点脂氧化抑制其激活
为深入解析15d-PGJ2抑制CaMKII的分子机制,作者分别采用生物素抗体和CaMKII抗体进行pull-down实验,发现15d-PGJ2以剂量依赖性方式与CaMKII结合。之后,利用质谱分析发现,15d-PGJ2可通过脂氧化方式修饰CaMKII-δ9的C495位点。该位点位于CaMKII的聚合结构域,是其形成十二聚体结构并实现活化所必需。突变该位点后,15d-PGJ2不能与CaMKII结合,失去了抑制CaMKII活性的能力,证实该位点是其关键作用位点。进一步实验表明,CaMKII-δ9 C495位点的脂氧化修饰通过破坏其全酶组装来抑制激酶活性。
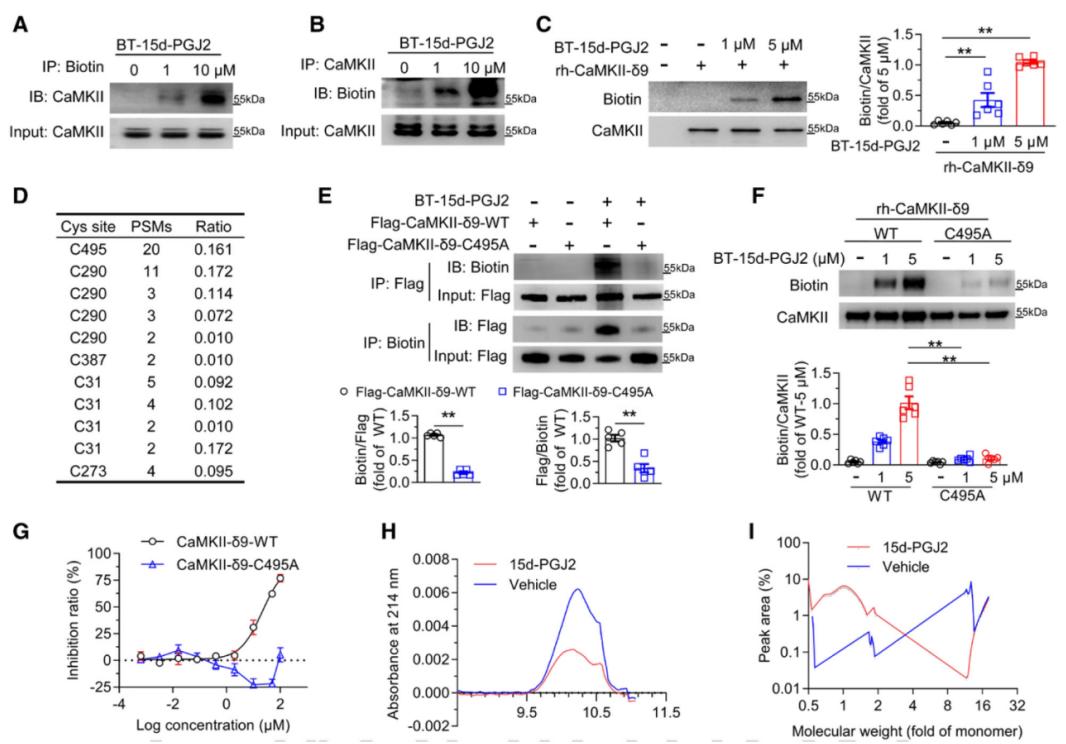
图8. 15d-PGJ2通过诱导CaMKII-δ9 C495位点脂氧化抑制其激活
总结
综上所述,在正常状态下,L-PGDS生成的15d-PGJ2通过脂氧化修饰抑制CaMKII活化,维持心肌细胞稳定存活;而在I/R损伤时,L-PGDS表达下调导致15d-PGJ2生成减少,CaMKII过度激活,最终引发心肌细胞死亡,加重心脏损伤。









 8434
8434










