2025年10月30日,广州医科大学第一附属医院曾国华教授团队等在《Molecular Cancer》(IF=33.9)发表了题为“Targeting CXCR2 in prostate cancer cells can block CD47-SIRPα interaction and reverse M2 macrophage polarization in the TME”的研究论文。该研究首次揭示IL-8/CXCR2通路可通过代谢重编程提升乙酰辅酶A(AC-CoA)水平,进而促进CD47乙酰化并上调其表达;同时诱导肿瘤细胞分泌棕榈酸,通过棕榈酰化修饰增强CD47膜定位,最终强化对巨噬细胞吞噬作用的抵抗。

值得注意的是,在本研究中,作者使用了汉恒生物提供的过表达和敲低细胞系: C4-2B/CXCR2 过表达、LNCaP/CXCR2过表达、C4-2B/IL-8过表达、PC3/CXCR2 敲低和PC3/CD47敲低,系统验证了上述机制。
神经内分泌前列腺癌(NEPC)具有显著的免疫逃逸特征,当前免疫检查点抑制剂疗法对其疗效有限,这一差距可能源于NEPC独特地依赖于脂质代谢驱动的免疫抑制,其中白细胞介素-8(IL-8)/C-X-C基序趋化因子受体2(CXCR2)信号轴已成为一个关键但尚未被充分探索的节点。其像藏在肿瘤微环境深处的一把“隐形钥匙”,虽尚未被完全拧动,却可能同时开启癌细胞转移、免疫逃逸与耐药的三重大门。最新证据表明,CXCR2信号可抑制CD47表达并促使巨噬细胞由M2表型重编程,提示该通路与吞噬免疫逃逸存在直接关联,但其具体机制尚未阐明。CD47,也被称为“别吃我”信号,在晚期前列腺癌症中升高,并受肿瘤微环境(TME)炎症变化的调节。然而,仅CD47上调并不能完全解释M2巨噬细胞极化,这突显了探索控制这一过程的其他代谢线索的必要性。
下面,我们一起来了解具体的研究内容:
1.IL-8/CXCR2通路调控肿瘤相关巨噬细胞(TAM)的浸润与极化
为了探索IL-8/CXCR2通路在TME中尤其是对巨噬细胞极化的作用,作者对临床组织样本进行了转录组测序,并分析比较了腺癌组和NEPC组之间的基因富集谱,发现NEPC组织中的CD47表达水平显著高于腺癌组织(图1A-D),免疫组化(IHC)染色验证了这一发现。NEPC细胞表现出高表达的IL-8、CXCR2和CD47,同时TAM的浸润减少,相比之下,腺癌样本中CD8+T细胞浸润更明显,且TAM数量更多(图1E-F)。为探究IL-8/CXCR2信号通路的作用,作者使用几种前列腺癌细胞系过表达CXCR2和IL-8。检测结果显示,CXCR2和IL-8仅在去势抵抗性前列腺癌(CRPC)和NEPC细胞系中存在(图1G)。此外,共培养实验验证了过表达CXCR2/IL-8可诱导条件培养基中巨噬细胞M2极化标志物CD206表达上调(图1H)。这些结果揭示了IL-8/CXCR2信号轴在调节前列腺癌微环境中的TAM浸润和极化中起着关键作用。

图1. IL-8/CXCR2通路调控TAM的浸润与极化
2.IL-8/CXCR2通路抑制巨噬细胞的迁移与吞噬功能
为进一步验证上述结果,作者通过体内实验发现,激活IL-8/CXCR2信号可促进肿瘤生长(图1A-C),流式和Transwell实验证明,该通路激活后肿瘤中巨噬细胞浸润减少,且吞噬活性(图D)和迁移能力也显著下降(图E-F)。向NOD-SCID伽马(NSG)小鼠外源性给予人IL-8,IL-8与其受体CXCR2的协同作用,显著削弱了肿瘤对卡马西平(PBMC)治疗的反应(图2G-H)。体外共培养模型中也观察到一致的治疗趋势(图2I)。对荷瘤小鼠模型组织进行的IHC分析显示,CXCR2表达升高与M1型巨噬细胞标志物减少、M2型标志物CD206增加相关(图2J-K)。Transwell迁移实验进一步证实,CXCR2激活能有效抑制TAM的迁移(图2L-M)。此外,经IL-8处理的荷瘤小鼠,TAM数量减少、吞噬功能下降,同时伴随M1型浸润减少与M2型浸润增加(图2N-O)。

图2. IL-8/CXCR2通路抑制巨噬细胞的迁移与吞噬功能
3.IL-8/CXCR2通路重塑肿瘤细胞的代谢特征
作者推测IL-8/CXCR2信号通路可能通过调控CD47表达,抑制TAM的募集,增强肿瘤免疫逃逸能力。接下来作者对其中的机制做了深入研究,代谢质谱(MS)分析和油红O染色显示,在CXCR2激活的荷瘤小鼠模型中,脂质代谢呈现显著增强,尤其是AC-CoA水平显著升高(图3A-B)。C13标记的代谢通量分析和Seahorse XF实验表明,AC-CoA 部分来源于葡萄糖代谢(图3C),抑制CPT1A及阻断CXCR2通路都会降低线粒体活性(图3D)。为深入了解脂质代谢与糖酵解对CXCR2表达的影响,作者对数据库进行了生物信息学分析,结果显示,脂代谢相关酶及葡萄糖转运蛋白SLC2A1在CXCR2高表达组中显著上调(图3E), Transwell实验证明用IL-8处理的C4-2B/CXCR2细胞可促进巨噬细胞迁移,该效应在PDC-E2或CPT1A抑制剂处理后进一步增强(图3F-H),说明巨噬细胞向肿瘤相关表型的转变。同时,IL-8/CXCR2信号激活能显著提升信号调节蛋白α(SIRPα)表达(图3I),并增加AC-CoA水平,而CXCR2阻断可逆转此现象(图3J)。这些结果表明IL-8/CXCR2通路通过代谢重编程推动肿瘤进展。
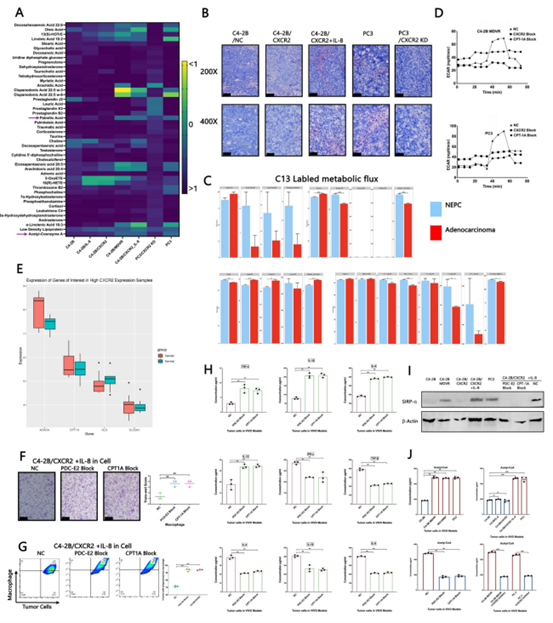
图3. IL-8/CXCR2通路重塑肿瘤细胞的代谢特征
4.IL-8/CXCR2通路通过p65乙酰化上调CD47表达
为进一步阐明IL-8/CXCR2调控CD47的机制,作者发现该通路激活可显著提高RelA/p65在赖氨酸310位点的乙酰化水平,并增加CD47表达(图4F-G)。棕榈酸处理同样增强p65乙酰化,提示脂代谢与NF-κB通路之间存在联系(图4H)。染色质免疫沉淀实验显示,K310R突变明显减弱了p65与CD47启动子的结合(图4I-K)。荧光素酶报告基因检测进一步证实,脂肪酸氧化激活剂或AC-CoA处理可显著增强NF-κB转录活性(图4A-B)。综上,IL-8/CXCR2诱导的代谢重编程通过提升乙酰辅酶A水平,促进p65乙酰化及NF-κB通路激活,从而上调CD47表达,增强肿瘤免疫逃逸。
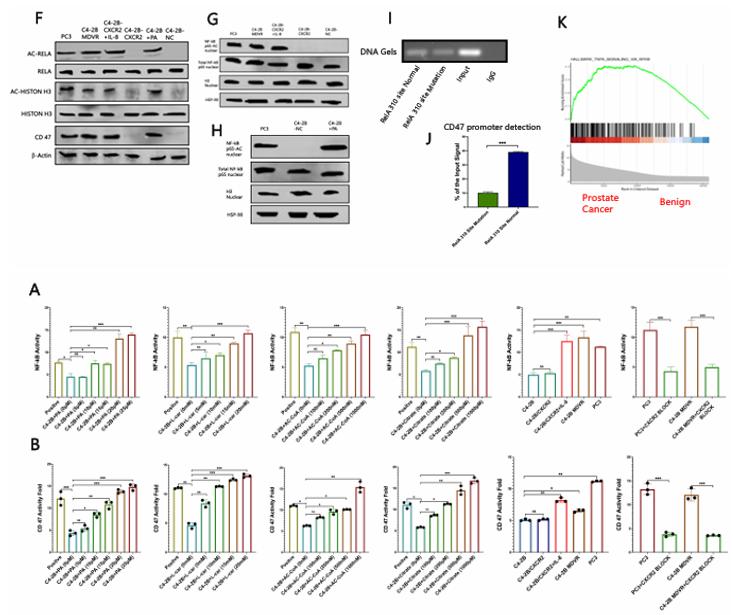
图4. IL-8/CXCR2通路通过p65乙酰化上调CD47表达
5.ZDHHC2介导的棕榈酰化调控CD47膜定位
接下来,作者使用Transwell实验发现棕榈酸处理的肿瘤细胞表现出更明显的抑制效果,继续研究S-棕榈酰化修饰(已知能调控蛋白质运输和膜定位的翻译后修饰),通过分析转染FLAG -CD47的前列腺癌细胞系PC-3和DU145,证实CD47存在棕榈酰化修饰(图5A-C)并借助CSS - Palm4.0预测工具锁定半胱氨酸33(C33)为关键修饰位点(图5D)。分子对接模拟显示,ZDHHC2是CD47结合亲和力最高的棕榈酰酰基转移酶(图5E),使用CRISPR介导的ZDHHC基因敲除和免疫共沉淀进一步确认了CD47与ZDHHC2存在直接作用(图5F-G),进一步实验表明,ZDHHC2过表达会增加CD47棕榈酰化水平和细胞膜上的定位,敲除则相反(图5H-I)。值得注意的是,全细胞裂解液中的总CD47水平则保持稳定(图5J-M)。通过与野生型CD47的对比发现,C33S突变体的细胞膜定位明显减弱(图5N-O),这些研究结果证实了ZDHHC2介导的CD47在C33位点的S-棕榈酰化修饰,是其定位于细胞膜的关键过程,这一机制对抑制巨噬细胞介导的抗肿瘤反应具有决定性作用(图5P)。
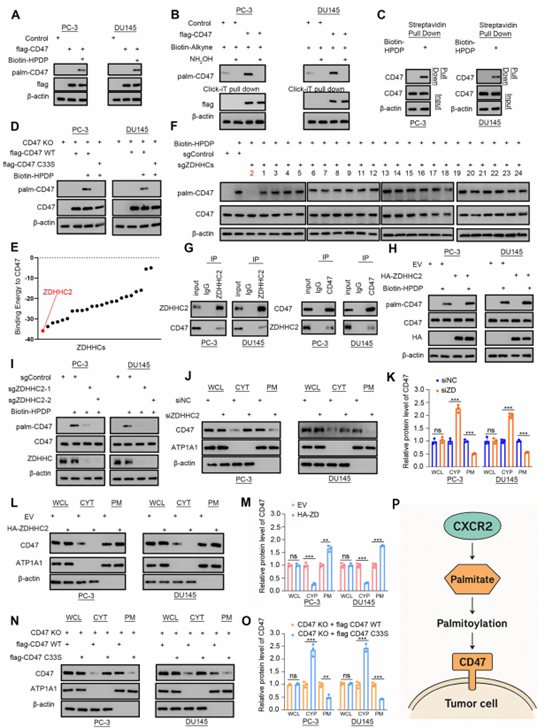
图5. ZDHHC2介导的棕榈酰化调控CD47膜定位
6.CD36高表达促进TAM向M2表型极化
为探究CD47高表达肿瘤微环境中巨噬细胞M2极化的原因,作者将过表达CXCR2的RM-1细胞原位植入C57BL/6小鼠的前列腺中(图6A), ELISA结果显示,TME内的CXCL15分泌随着肿瘤生长而显著增加,而细胞内CXCL15水平则保持稳定(图6B-C)。通过评估从脾脏和TME分离巨噬细胞的脂质摄取能力,发现在TME中TAM表现出更高的脂质摄取(图6D)。先前研究显示CD36是一种已知介导脂质摄取并促进M2极化的清道夫受体,作者使用脾源性巨噬细胞在体外与CXCL15共培养,观察到CD36表达和M2标志物上调(图6E)。阻断CXCR2或CD36显著减少了TAM对VLC -多不饱和脂肪酸的摄取(图6F-G),并抑制了M2相关细胞因子的分泌(图6H-I)。此外,使用共培养系统,抑制CD36能够显著降低M2极化(图6J-K),体外实验显示,ω-3和ω-6 VLC-PUFAs能够强烈的促进了M2极化(而棕榈酸作用有限)(图6L),且该效应可以被CD36抑制所消除(图6M-N)。这些发现揭示了信号轴在驱动TAM向M2极化中的关键作用。
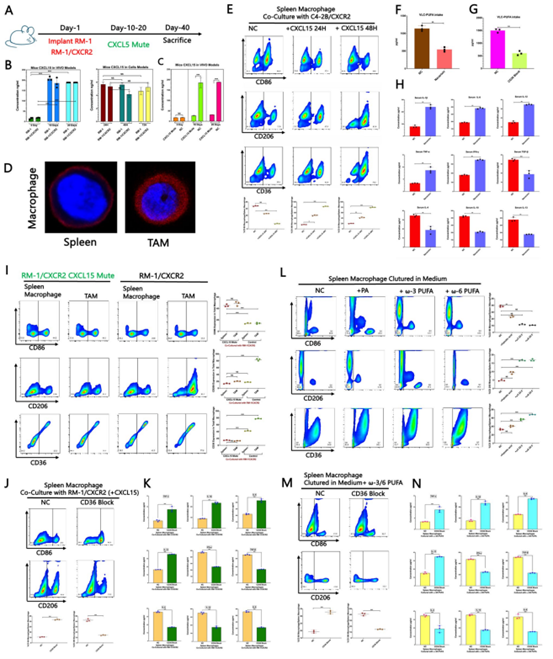
图6. CD36高表达促进TAM向M2表型极化
7.CXCR2抑制剂的体内抗肿瘤效果
最后,作者在两种小鼠模型中评估了CXCR2阻断的治疗潜力。在NSG小鼠模型中,使用纳瓦里辛(CXCR1/2拮抗剂)或血小板反应蛋白-1(TSP-1)治疗的荷瘤小鼠,其肿瘤体积显著缩小(图7A-B)。此外,抑制CXCR2后M1相关细胞因子增加,促进向促炎巨噬细胞表型转变(图7C),同时肿瘤相关巨噬细胞总体数量增加,对肿瘤细胞的吞噬活性受损(图7D-F)。在C57BL/6小鼠模型中也得到了类似结果(图7G)。综合来看,抑制CXCL15/CXCR2信号通路不仅能显著抑制肿瘤进展,还能在临床前模型中激发强烈的抗肿瘤免疫反应(图7I-J)。
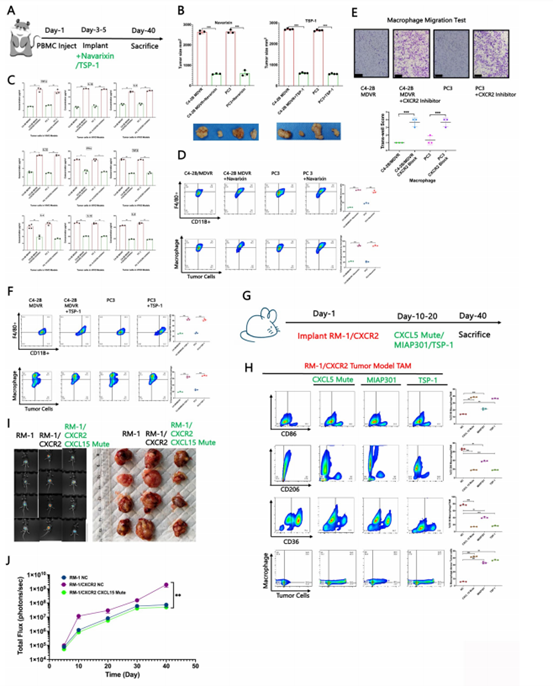
图7. CXCR2抑制剂的体内抗肿瘤效果
总结
本研究揭示了IL-8/CXCR2信号轴在前列腺癌免疫逃逸中的核心作用。该通路通过代谢重编程上调AC-CoA水平,促进p65乙酰化,从而转录激活CD47。同时,经由ZDHHC2介导的棕榈酰化修饰,CD47被稳定于细胞膜,增强其“别吃我”信号。此外,该轴还通过分泌脂质诱导巨噬细胞向M2型极化,协同抑制吞噬功能。因此,靶向CXCR2能同时阻断CD47-SIRPα互作并逆转M2极化,为增强抗肿瘤免疫与克服当前免疫治疗耐药提供了新的潜在策略。









 8434
8434










