2025年12月,徐州医科大学吕凌团队联合南京医科大学第一附属医院古鉴团队在《Cell》期刊上发表了题为“Tumor-produced ammonia is metabolized by regulatory T cells to further impede anti-tumor immunity”的研究论文。该研究深入揭示了肿瘤微环境中调节性T细胞(Treg)通过代谢氨增强其免疫抑制功能的分子机制,为理解肿瘤免疫逃逸及免疫治疗耐药提供了新视角。值得注意的是,文章中使用了汉恒生物提供的pGL3-SMS-promoter和pcDNA3.1-FOXP3质粒来研究FOXP3和SMS基因之间的调控关系,为结论提供了关键实验支持。

在肿瘤进展过程中,Tregs会在肿瘤微环境(TME)中逐渐积累并抑制抗肿瘤免疫反应,而效应T细胞的数量则相应减少。Tregs通过何种机制来适应不断恶化的TME,一直是肿瘤免疫治疗领域的未解之谜。肝细胞癌(HCC)是一种高度恶性的肿瘤,其对抗程序性细胞死亡蛋白1(PD-1)/抗程序性死亡配体1(PD-L1)治疗的应答率低于20%。肝脏是氨代谢的主要场所,因而作者推测Tregs可能通过适应重塑的氨环境来抑制肿瘤免疫反应。
本研究通过空间代谢组学与转录组学分析发现,人类HCC中存在代谢异质性亚区,这些区域谷氨酰胺分解代谢活跃,氨水平升高,且Tregs富集,而效应T细胞(CD8⁺T和CD4⁺T)则出现死亡。机制上,一方面Tregs通过上调精氨基琥珀酸裂解酶(ASL),启动尿素循环来消除氨的毒性;另一方面,氨还能在FOXP3转录因子调控的精胺合成酶(SMS)作用下转化为精胺。X射线晶体学证实,精胺与过氧化物酶体增殖物激活受体γ(PPARγ)直接结合,进而全面调控多种线粒体复合体蛋白的转录,以增强Tregs的氧化磷酸化(OXPHOS)和免疫抑制功能。临床层面,经抗PD-1治疗后死亡的肿瘤细胞通过转氨脱氨作用释放氨,进而增强Treg功能,最终导致免疫治疗耐药。因此,靶向氨生成以抑制Tregs,有望成为抗肿瘤免疫治疗的潜在策略。
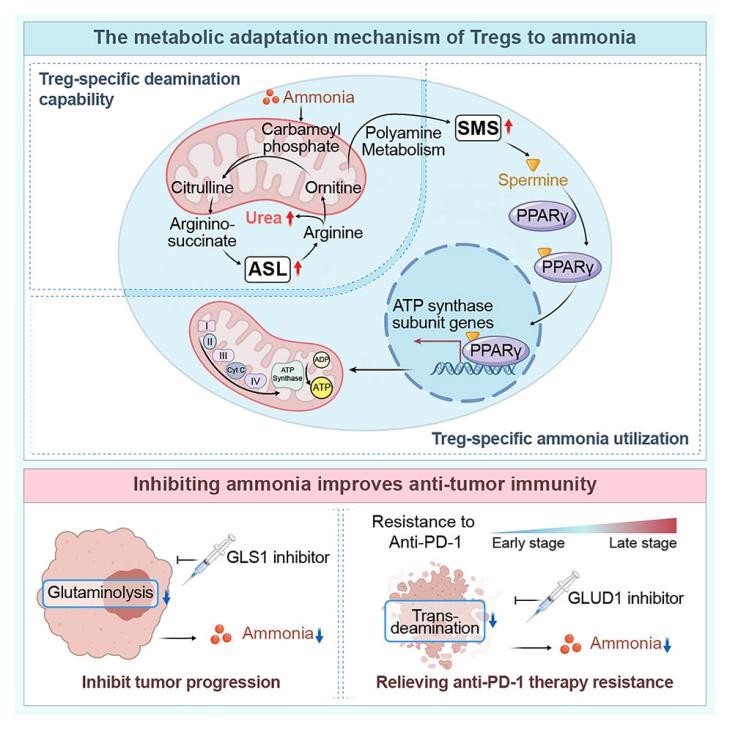
图1.Treg适应肿瘤氨代谢的分子机制
下面,我们一起来了解具体的研究内容:
1. 高谷氨酰胺代谢-低尿素循环的肿瘤亚区富集Treg
为了阐明肿瘤区域代谢特征与免疫细胞分布之间的关系,作者首先对6例肝癌患者的组织样本进行了空间转录组和空间代谢组分析(图2A),发现Treg主要分布在肿瘤组织,其他免疫细胞则主要集中在癌旁组织(图2B-C)。通过分析不同免疫细胞浸润区域(尤其是Treg区域)的能量代谢特征,并结合KEGG分析结果,作者发现不同T细胞亚型的分布区域具有独特的代谢特征,肿瘤中Treg富集区域表现出一种独特的代谢特征:高度活跃的谷氨酰胺分解和显著降低的尿素循环活性(图2D-H)。在小鼠肝癌模型中,这一空间相关性和代谢特征得到了进一步验证:随着肿瘤进展,谷氨酰胺分解活性升高,瘤内氨浓度增加,Treg丰度同步增加。
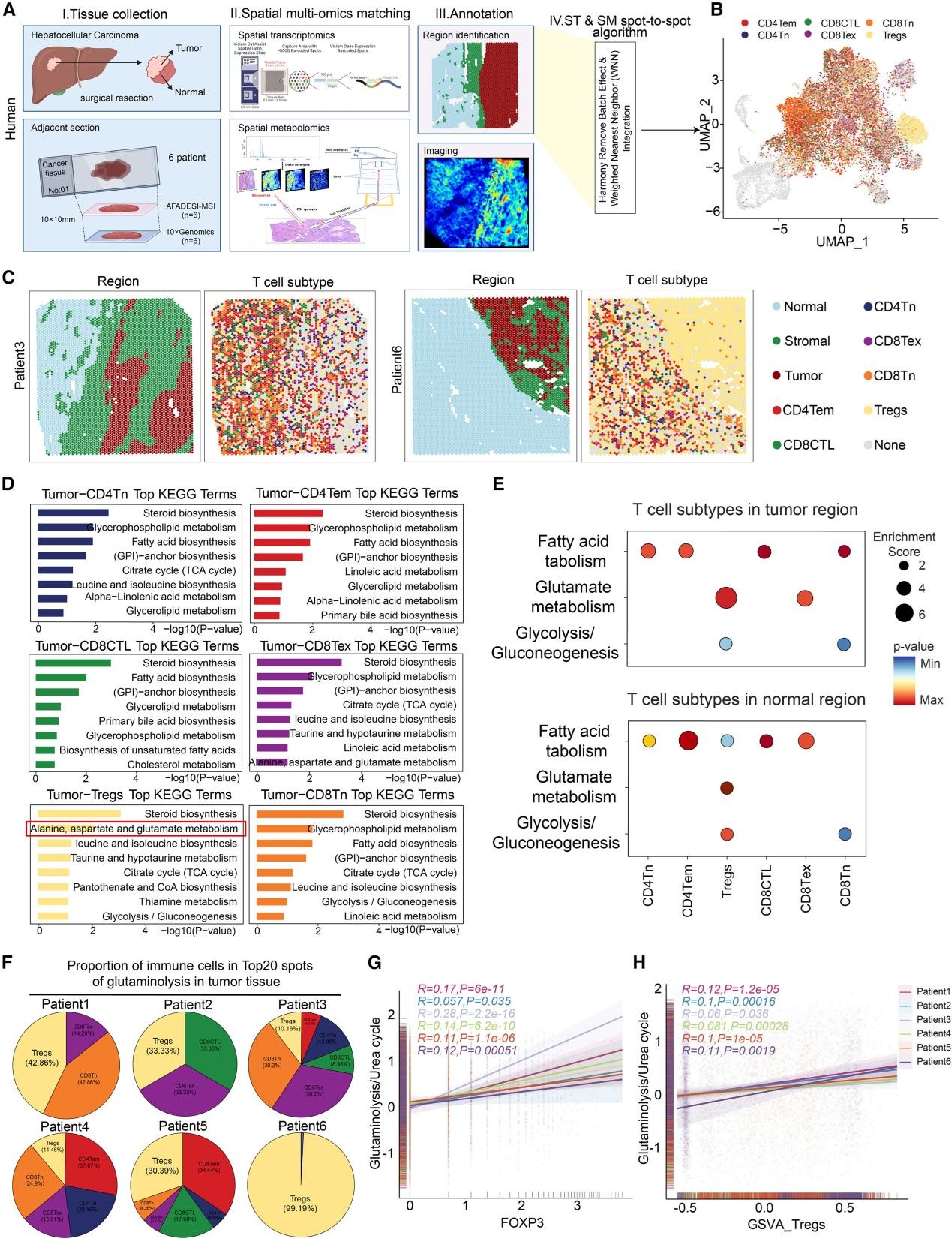
图2. 谷氨酰胺分解代谢活跃和尿素循环活性降低是肿瘤中Treg富集区域的关键代谢特征
谷氨酰胺分解的主要副产物是氨,而尿素循环是清除氨的主要途径,二者共同造就了一个高氨的环境。作者猜测,局部氨积累可能是驱动Treg富集的关键因素,并利用体内外实验进行验证,发现在尿素循环活性相对较高的肿瘤样本中,氨水平较低,Treg富集也较弱(图3J)。体内实验也显示,肿瘤内注射氨会促进肿瘤生长并提高Treg富集(图3K-L)。
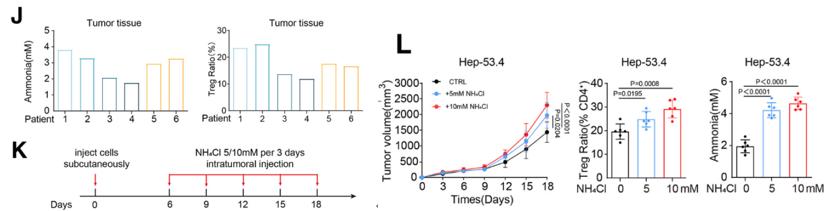
图3. 氨促进肿瘤生长并提高Treg富集
2. Treg而非CD8+T或CD4+T细胞可抵抗氨诱导的细胞凋亡
那为何氨会选择性地“偏爱”Treg呢?作者将从健康外周血单个核细胞(PBMCs)中分离出的Treg、CD8+T和CD4+T细胞置于不同的NH4Cl浓度环境中进行实验,发现与肿瘤内浓度相当的氨(5mM)几乎不影响Treg的增殖,却显著抑制了CD8+T和CD4+T细胞的增殖,高浓度的氨(10mM和20mM)会抑制所有细胞增殖,对CD8+T和CD4+T细胞影响更明显(图4A)。流式细胞术和蛋白印迹结果显示低氨(5mM)环境下,CD8+T和CD4+T细胞会发生细胞凋亡,而Treg则表现出抵抗凋亡的能力(图4B-C)。此外,低浓度氨处理后的Treg,其关键转录因子FOXP3和肿瘤免疫抑制性分子PD-1、CTLA-4的表达上调,免疫抑制功能增强(图4D-E)。动物实验结果与细胞实验一致,高氨饮食的小鼠脾脏和外周血中的Treg丰度增加,CD8+T和CD4+T细胞丰度降低,注射鸟氨酸(可减少氨的积累)则能够逆转这些效应(图4F-J)。以上数据表明Treg在氨胁迫下具有明显的生存和功能优势,而效应T细胞则高度敏感。
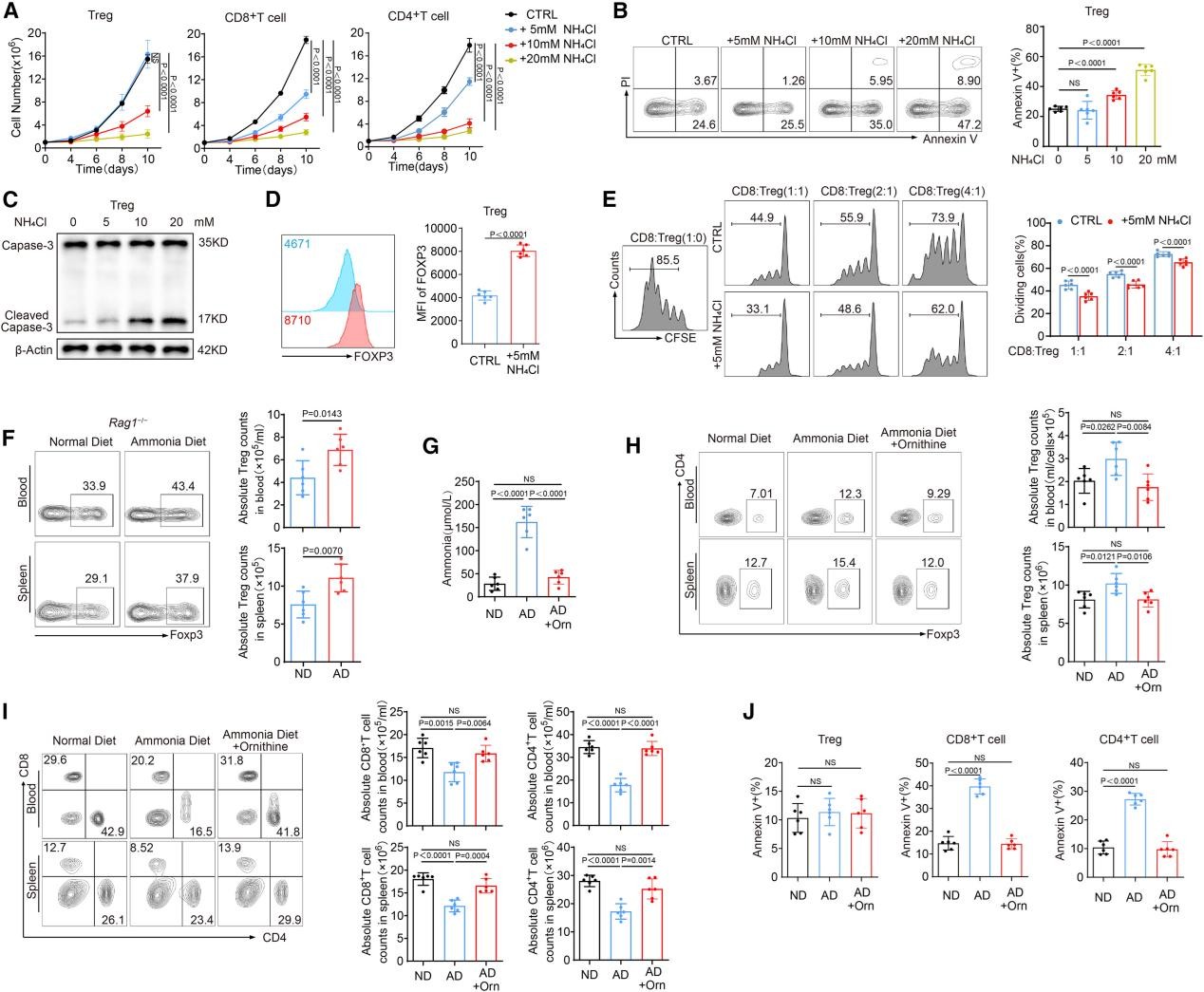
图4. Treg能够抵抗氨诱导的细胞凋亡
3. 氨通过上调ASL的表达激活Treg中的尿素循环
基于以上实验结果,作者提出假设:Treg在氨代谢方面比其他T细胞亚群表现得更为出色。为验证这一假设,作者从HCC样本中分离出Treg、CD8+T细胞和CD4+T细胞检测。发现相比CD8+T和CD4+T细胞,Treg中的氨水平较低,且存在大量的尿素循环中间产物(图5A-B)。利用15N标记的氨进行示踪实验,发现Tregs中尿素循环中间产物的同位素掺入量更高(图5C-D)。这些结果表明在富氨环境中,Treg具有较强的尿素循环活性。
氨是如何驱动Treg的尿素循环的呢?带着这个疑问,作者分析了HCC的单细胞RNA测序(scRNA-seq)数据,发现肿瘤浸润的Treg会特异性地高表达尿素循环中的一个关键酶——ASL(图5E)。使用5mM的NH4Cl处理Treg,结果显示ASL的mRNA和蛋白水平均上调,与scRNA-seq结果一致,说明氨通过上调ASL来诱导Treg中尿素循环的激活。ASL敲低后,Treg中氨含量升高,增殖能力和抗凋亡能力减弱,尿素循环能力受损(图5I-N)。这揭示了Treg感知和适应氨压力的第一道防线:ASL介导的尿素循环激活。
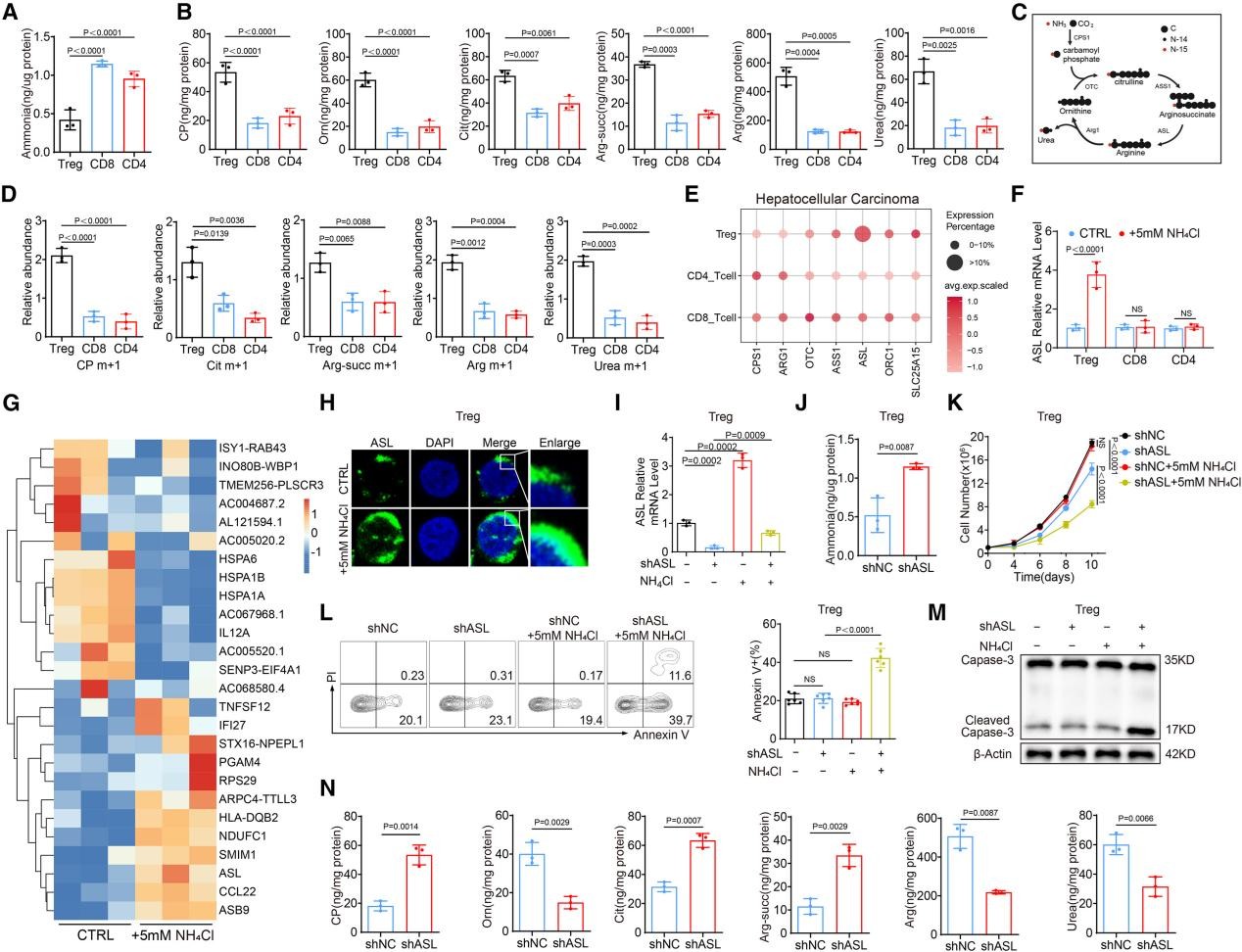
图5. 氨通过上调ASL的表达激活Treg中的尿素循环
4. 氨通过SRC3介导的STAT3激活作用,增强Treg中的ASL转录过程
接着,作者进一步探究ASL的上游通路,通过分析氨处理Treg的RNA测序(RNA-seq)数据,发现信号转导及激活转录因子3(STAT3)可激活ASL表达,药物抑制或者敲低STAT3均会抑制氨对ASL的上调作用(图6N-P)。转录因子与启动子的双荧光素酶报告基因实验验证STAT3作为转录因子与可结合在ASL的启动子区域来激活其转录,结合位点为ASL启动子上游263-271bp(图6S-U)。通过免疫沉淀实验,发现类固醇受体共激活因子3(SRC3)是氨诱导的STAT3共激活因子中的重要成员(图6V)。单细胞分析显示,SRC3的表达与肿瘤内Treg中的抗原呈递淋巴小结(ASL)呈特异性且正相关(图6W至S3Y)。氨诱导能够增强Treg中STAT3-SRC3复合物形成,激活ASL转录,降低细胞凋亡,敲低SRC3后上述效应会受到抑制,过表达野生型SRC3后可恢复这些效应(图6AB至S3AE)。
综上,氨促进了SRC3与STAT3的相互作用,从而驱动ASL的转录过程,这一机制解释了Treg在TME中感知和适应氨压力的独特能力。
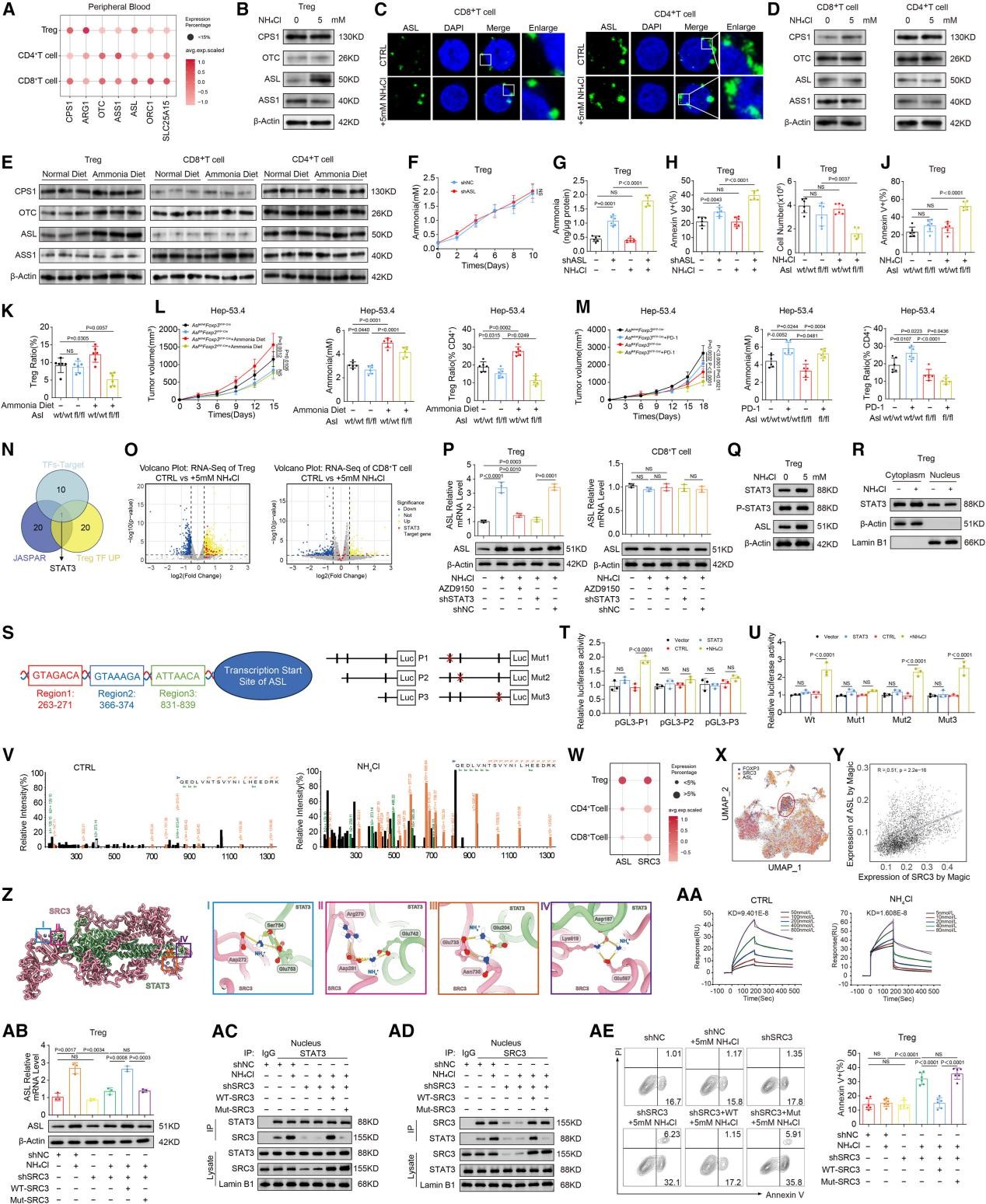
图6. 氨通过上调ASL表达激活Treg的尿素循环
5. 氨通过由FOXP3调控的SMS表达,促进Treg中的OXPHOS
氨含量丰富的肿瘤微环境中,Treg的OXPHOS通路显著富集,线粒体功能检测也证实,氨处理提升了Treg的耗氧率(OCR)、ATP产量和线粒体膜电位,而CD8+T和CD4+T细胞则没有这种变化(图7A-F)。氨能够诱导Treg中FOXP3上调,增强Treg中肿瘤免疫抑制分子的表达,然而这些效应均会被OXPHOS抑制剂消除,说明OXPHOS的增强对于维持FOXP3表达和Treg的肿瘤免疫抑制功能至关重要。
利用液相色谱-串联质谱法(LC-MS/MS)技术,作者发现HCC和Treg中与氨代谢相关的SMS基因上调,与肿瘤来源Treg中精胺水平升高现象一致(图7G-I)。敲低Treg中的SMS后,会抑制OCR的增强、破坏线粒体的完整性,并抑制精胺的产生(图7J-K),另外,敲低SMA还会导致OXPHOS减弱,线粒体基因表达下调,免疫抑制功能受损(图7L-N)。根据JASPAR和PROMO数据库预测结果,FOXP3是SMS的潜在调节因子,利用转录因子与启动子的双荧光素酶报告基因实验,作者确认FOXP3是SMS的转录激活因子,染色质免疫共沉淀(ChIP)-qPCR和电泳迁移率变动分析(EMSA)的结果与双荧光素酶报告基因实验结果一致(图S4O-V)。这些结果确立了FOXP3作为驱动SMS表达的转录激活因子,增强了氨诱导的Treg的OXPHOS水平。
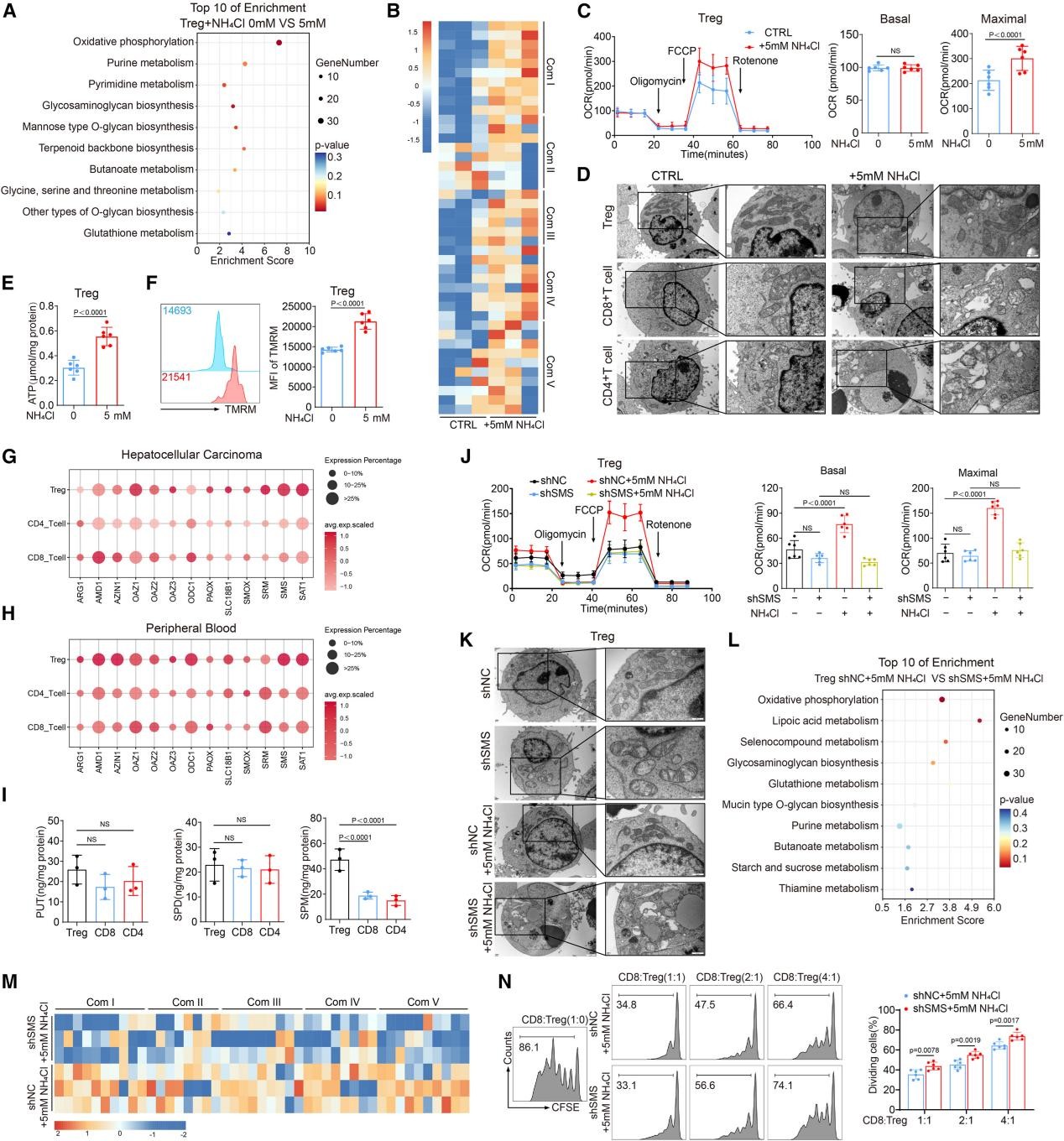
图7. 氨通过FOXP3-SMS轴促进Treg的OXPHOS
6. 精胺通过PPARγ促进Treg的OXPHOS并增强其免疫抑制能力
SMS能促使精胺降解转化为精氨酰胺,作者在SMS敲低的Treg中补充精胺,发现当精胺转化为精氨酰胺后,显著增强了Treg的OXPHOS水平(图8A-C)。过表达精胺/精胺N(1)-乙酰转移酶SAT1(精胺降解的限速酶)后,精胺、精氨酰胺、OXPHOS水平均下降(图8D-F)。表明精氨酰胺是驱动氨诱导条件下Treg发生OXPHOS的关键代谢物。
精氨酰胺如何增强OXPHOS?为阐明这一机制,作者开发了精胺衍生物探针SPM-DA,通过蛋白质组学技术,结合液相色谱-质谱分析、反向靶标筛选和基于RNA-seq的转录因子预测,作者锁定了一个关键靶点:过氧化物酶体增殖物激活受体γ(PPARγ)(图8G-N)。NH4Cl处理Treg后PPARγ的表达未出现明显变化。然而,在Treg中敲低PPARγ会抑制NH4Cl诱导的高OCR(图8O-P)。Treg特异性PPAR-γ敲除(Pparγfl/flFoxp3YFP-Cre)小鼠的氨诱导实验和皮下成瘤实验进一步验证,NH4Cl或内源性精胺通过靶向PPARγ来增强Treg的功能。
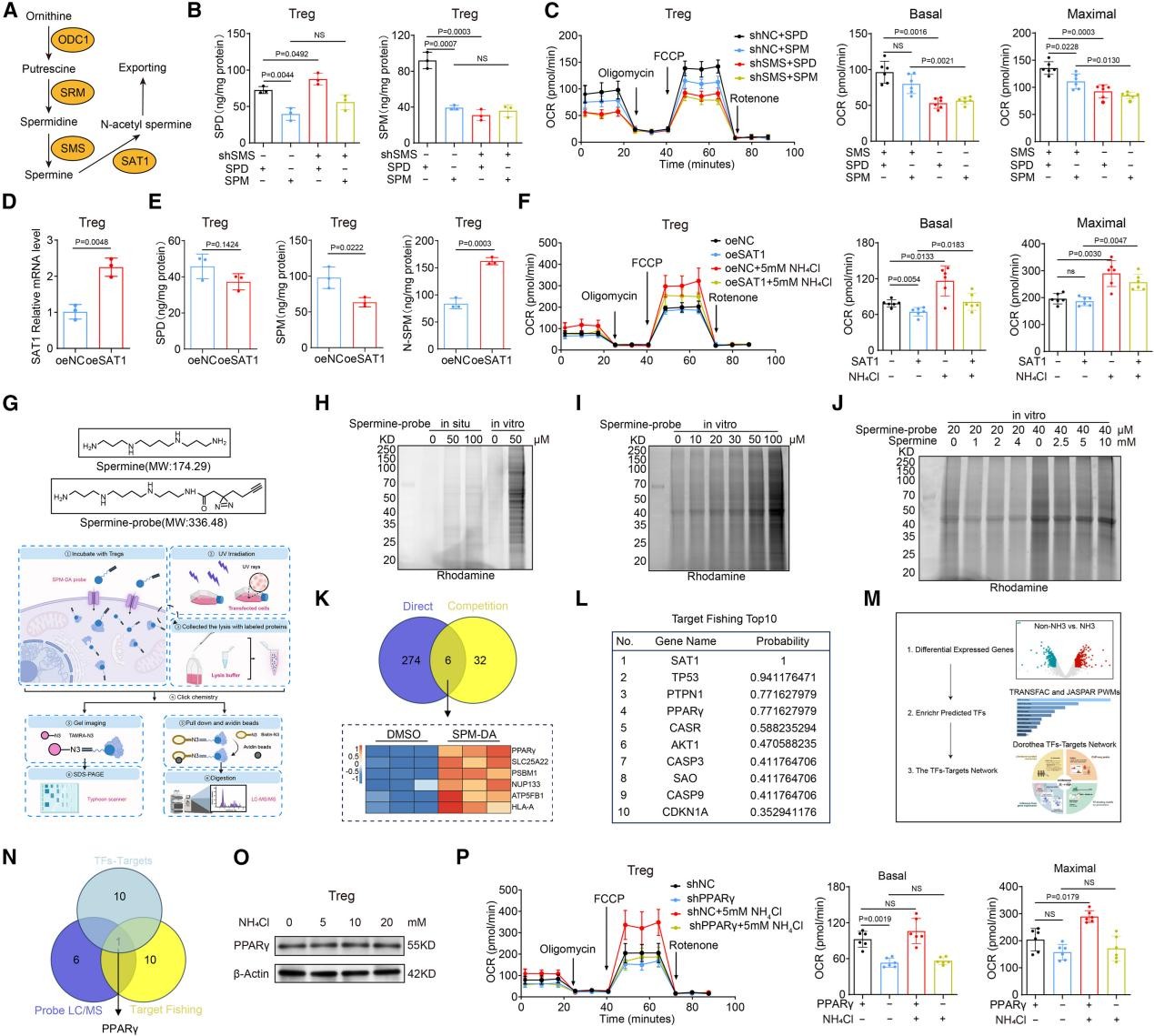
图8. 精胺通过PPARγ增强Treg的OXPHOS和免疫抑制能力
作者利用微尺度热泳分析(MST)、等温滴定热分析(ITC)、细胞热位移测定法(CETSA)和药物亲和响应靶稳定性(DARTS)验证了精胺与 PPARγ之间存在直接相互作用(图9A-D)。通过X射线晶体学、分子对接和突变分析,作者精确定位了精胺与PPARγ结合的关键氨基酸位点Glu319, Ser370, Glu371(图9E-H),将这些位点突变为Ala,生成PPARγ-MUT蛋白,有效地消除了与精胺的结合(图9I-6L)。PparγMutFoxp3YFP-Cre小鼠的CETSA和DARTS实验结果显示Treg中PPARγ与精胺的结合稳定性显著降低(图9M-N)。此外,精胺-PPARγ复合物通过上调多个线粒体呼吸链复合物的基因表达,增强了Treg的OXPHOS。
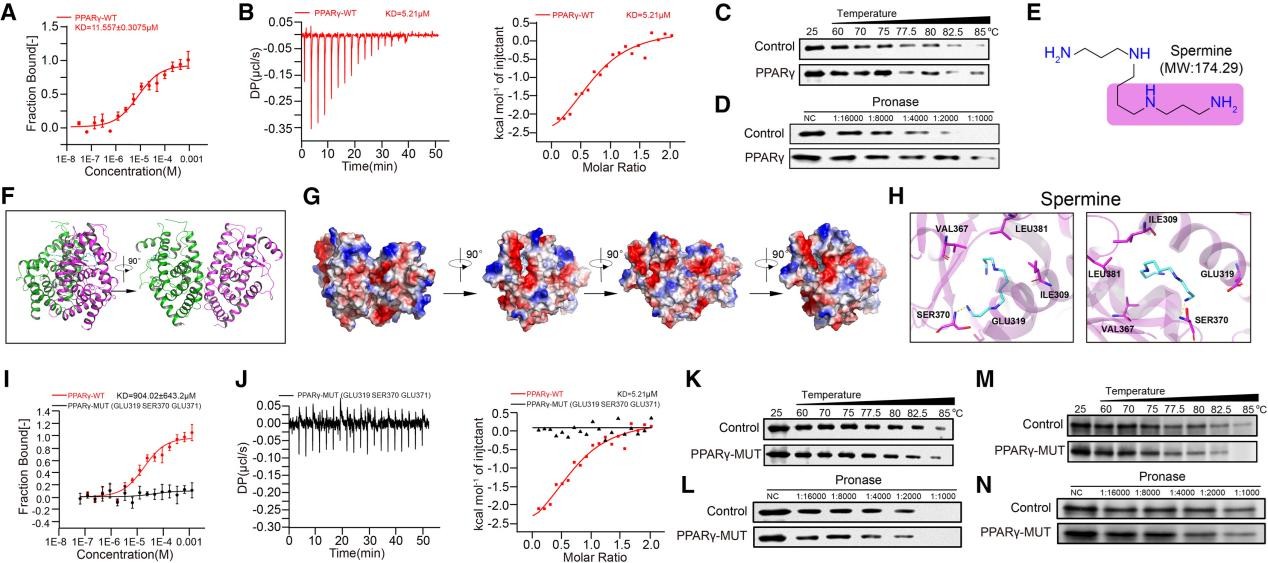
图9. 精胺与Treg中的PPARγ直接结合
. 抗PD-1疗法诱导的细胞死亡所产生的氨会增加Treg的数量并导致免疫治疗耐药
临床数据显示,使用抗PD-1/PD-L1进行免疫治疗后,Treg数量增加。因此作者推测免疫治疗可能会促进肿瘤内部Treg的富集,接受抗PD-1治疗的HCC患者样本证实了这一猜测,组织中氨含量升高,Treg浸润增加(图10A)。在多种肿瘤细胞系中诱导细胞凋亡的结果表明,细胞死亡会提高上清液中的氨浓度,代谢组学分析显示,这种凋亡相关的氨主要来源于转氨脱氨作用(而非谷氨酰胺分解),即谷氨酸在谷氨酸脱氢酶GLUD1作用下生成α-酮戊二酸并释放氨,临床抗PD-1治疗样本的分析结果与细胞一致(图10B-D)。
在Hep-53.4细胞皮下移植肝癌小鼠模型中,抗PD-1治疗初期能抑制肿瘤生长,但随后出现耐药,并伴随着瘤内氨和Treg水平升高。此时,联合使用谷氨酰胺酶抑制剂CB-839无法克服耐药,但联合使用GLUD1抑制剂R162却能有效控制肿瘤的再生长,有效地降低了肿瘤内的氨水平,Treg数量减少(图10E-G)。PD-1耐药的肿瘤中α-KG水平升高,这反映了转氨脱氨基作用的激活,而这一现象通过谷氨酰脱氢酶抑制得以逆转(图10H)。相比之下,谷氨酸水平相对保持不变(图10I)。在Hep-53.4细胞原位肝癌小鼠模型中也观察到了类似的效果。R162联合抗PD-1治疗能有效抑制肝癌进展,并降低肿瘤内氨水平和调Treg浸润,与仅使用R162的组相比治疗效果更为显著(图10J–N)。
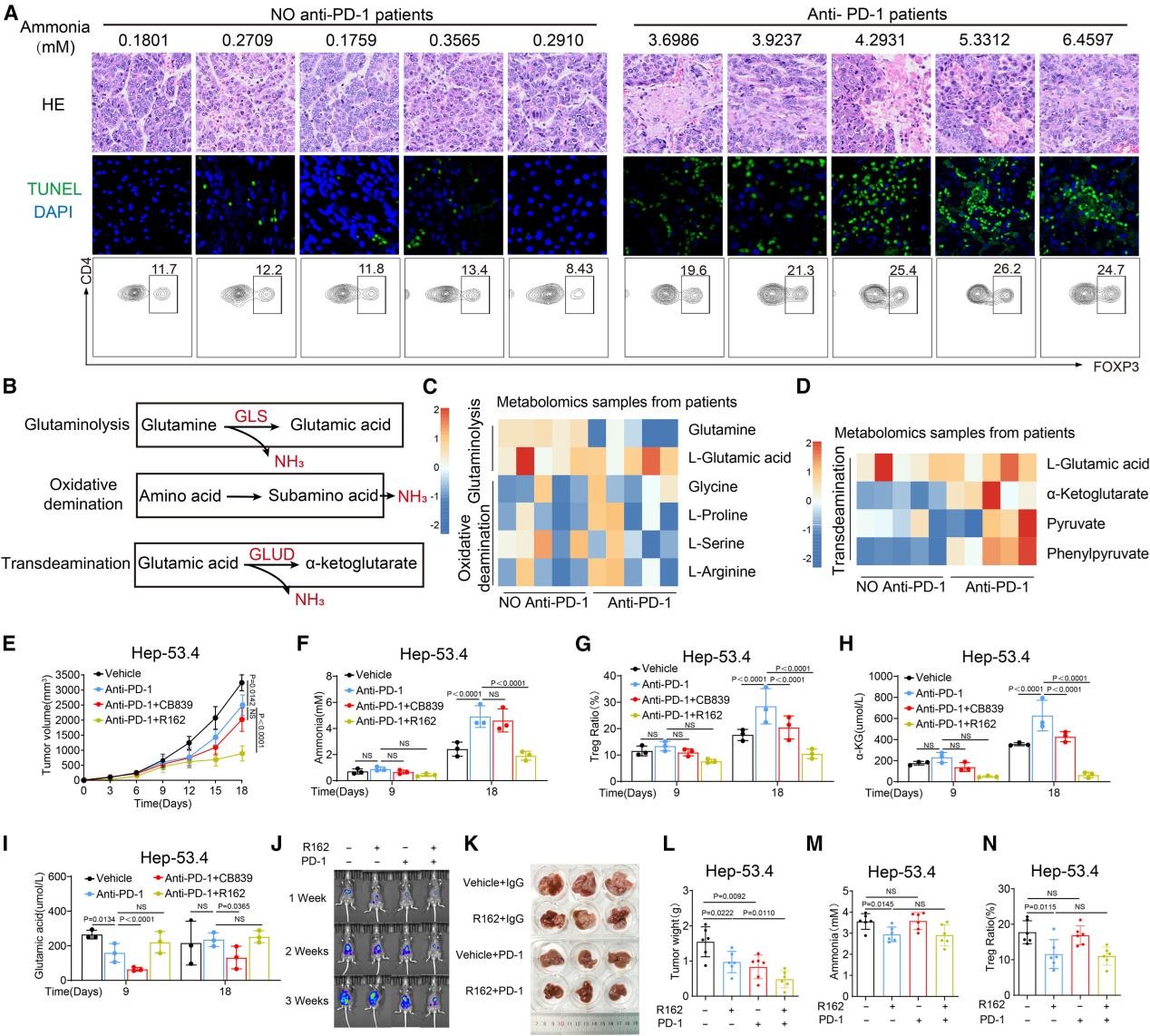
图10. 抗PD-1疗法诱导的细胞死亡所产生的氨会增加Treg的数量并导致免疫治疗耐药
综上,该研究首次阐明了氨在Treg适应性免疫抑制中的核心作用,靶向肿瘤的氨生成可以有效降低肿瘤微环境氨水平、削弱Treg功能,从而与现有免疫疗法产生协同作用,为提出了潜在的联合治疗方案奠定了坚实的理论基础,为理解肿瘤免疫逃逸机制提供了新视角,也为抗肿瘤免疫治疗提供了新靶点。









 8152
8152










