在上一期的干货文章中,我们系统的介绍了炎症小体与焦亡的关系,本期内容我们将介绍焦亡通路中的另一个重要角色——NF-κB
NF-κB 是一组蛋白质复合体的统称,主要包含 RelA (p65)、RelB、c-Rel、p50/p105 (NF-κB1) 和 p52/p100 (NF-κB2) 等亚单位。NF-κB发现于1986年的B淋巴细胞的研究中,广泛分布于心肌细胞、血管内皮细胞、免疫细胞和神经元等多种细胞类型中。作为细胞内主要的转录因子,NF-κB能对细胞因子、生长因子、病原体感染、辐射和应激等多种细胞外刺激作出反应。NF-κB激活后会迅速进入到细胞核与靶基因启动子结合,调节基因的转录,从而影响细胞增殖、分化、凋亡、基因组稳定性和氧化应激等过程。此外,它还在炎症、免疫反应中发挥重要作用,对人体免疫反应、细胞死亡以及肿瘤的发生发展发挥重要的作用。

NF-κB的结构
NF-κB是一种在真核细胞中由Rel蛋白家族的多个亚基 (RelA (p65), c-Rel, RelB, NF-κB1 (p50), and NF-κB2 (p52))组成的转录因子,其中 p65/p50形成的异二聚体是哺乳动物细胞中最为常见的形式。这些Rel蛋白在N端有一个保守的RHR序列(由NTD和CTD构成),在CTD下游通常存在个一NLS序列,RHR序列具有DNA的结合、二聚体化和核转移的功能。
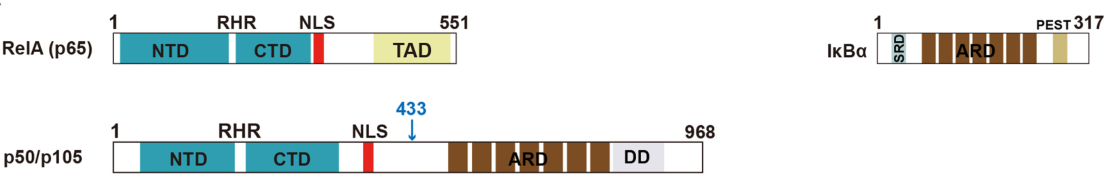
Rel蛋白结构域示意图
RelA、c-Rel和 RelB能直接和DNA结合,其C端存在一个反式激活结构域,该结构域可以用于激活基因的表达。与其他家族成员不同,p50 和 p52 分别来自于更大的前体 p105 和 p100。虽然p50和p52缺乏反式激活结构域,但是具有抑制基因表达的功能。除了P50和P52序列外,p105 和 p100还包括IkB样的锚蛋白区,它抑制与其相关的NF-kB亚单位的活性。从前体产生P50和P52的过程还没有被完全阐明,但他需要翻译时和翻译后的蛋白酶的加工。
NF-κB 主要以两种二聚体形式存在:一种是与NF-κB抑制单元( NF-κB inhibitory units)结合,如IκBα;另一种是结合DNA。NF-κB抑制单元通常是IκB的家族成员,主要包括p100, p105, IκBα, IκBβ, IκBγ, IκBε, Bcl-3, IκBNS, 和 IκBζ。这些家族成员具有一些共同的特征,包括N端的信号接收区域,锚定位点区域,富含脯氨酸、谷氨酸、丝氨酸和苏氨酸的CTD区域,以及能诱导IκB降解的泛素化位点。在静息状态下,IκBα通过自身的锚定序列与NF-κB结合,掩盖了NF-κB的NLS序列,从而限制了NLS的入核功能。
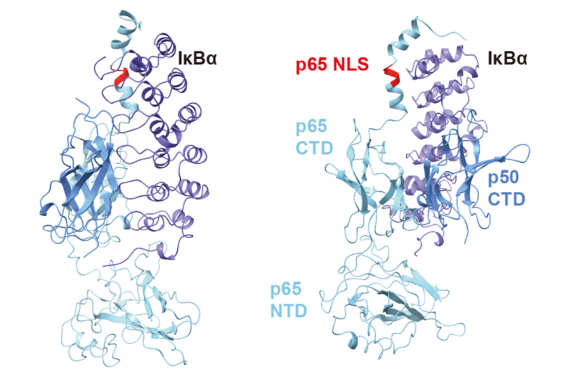
IκBα的三维结构
NF-κB 是一种能与DNA结合的蛋白,其能通过形成不同的二聚体,进而选择不同的DNA序列,调节不同基因的表达。两个RHR结构域会形成一个蝴蝶状的结构,中间位置能识别结合十个碱基左右的DNA序列。到目前为止,已经发现NF-κB 可调控200多个靶基因,其中大部分参与免疫和炎症反应。
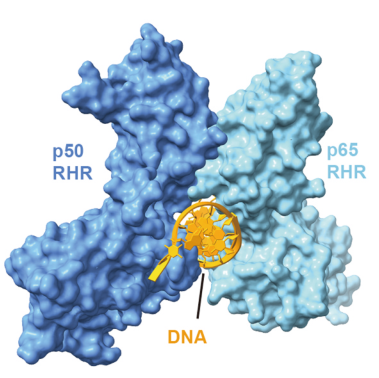
NF-κB 结合DNA的三维结构
NF-κB信号通路的激活
NF-κB信号通路的激活过程主要有两种途径:经典途径和非经典途径。在经典通路中, IκB 激酶的降解是核心步骤,NF-κB 二聚体通过 IKK 介导的 IκB 磷酸化进行激活,磷酸化过程会引起蛋白酶体 IκB 的降解,IκB的降解会导致p65/p50二聚体的释放,这使得活性 NF-κB 转录因子亚基易位到细胞核并诱导靶基因的表达。非经典路径依赖于RelB和p50的加工来激活整个信号通路。这两种途径都广泛参与炎症和免疫的生理过程。
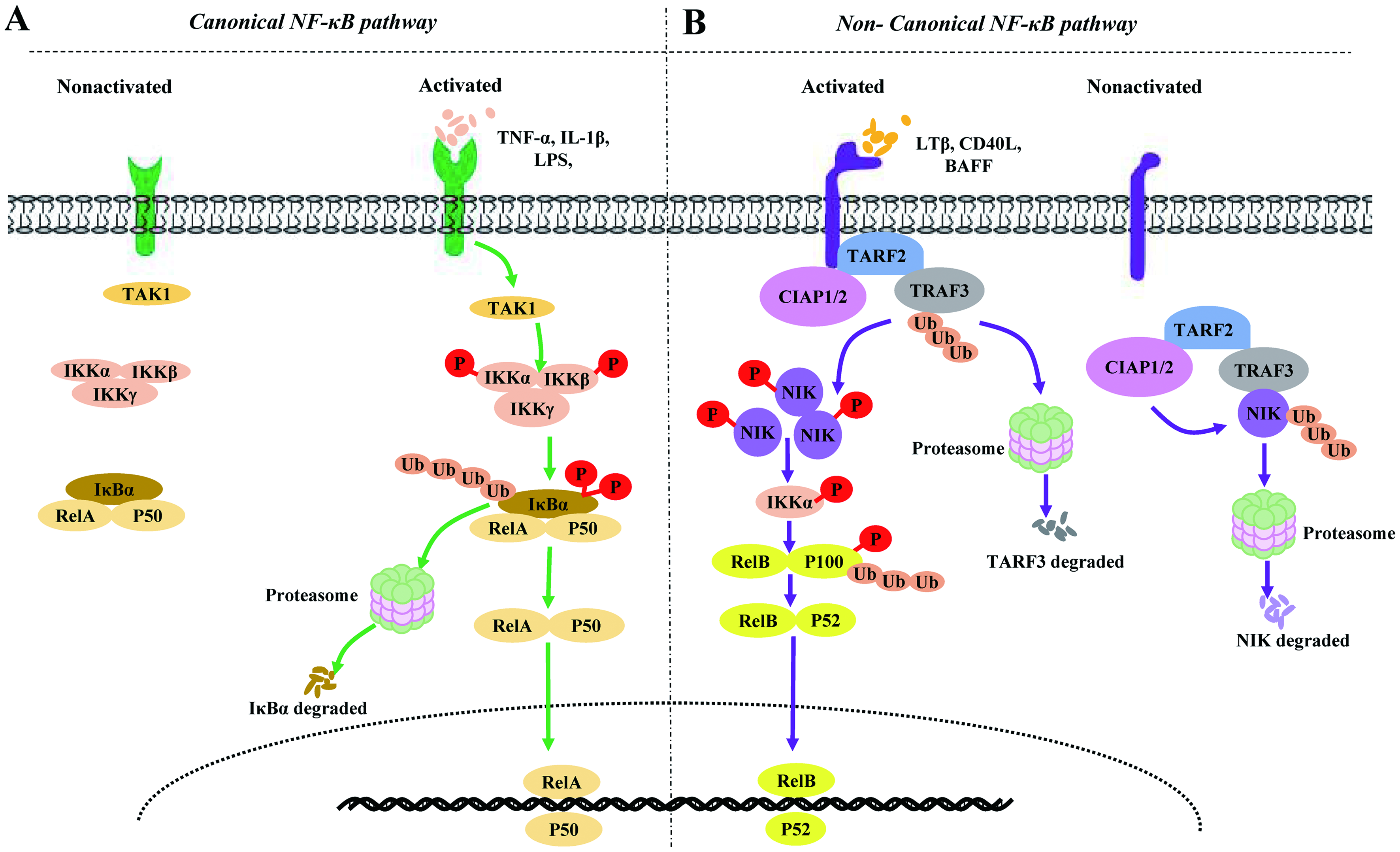
NF-κB信号通路
在经典路径中,NF-κB可响应多种信号,包括细胞因子受体、TLRs、模式识别受体(pattern recognition receptors)、肿瘤坏死因子受体超家族、t细胞受体、b细胞受体等。上游的刺激,如炎症、缺氧、机械刺激等,最终信号会汇集到IKK蛋白上,激活后的IKK能作用NF-κB-IκB 复合体上IκB亚基的丝氨酸位点,使其发生磷酸化,进而使IκB发生泛素化降解,NF-κB二聚体被抑制的状态解除,核定位序列暴露,NF-κB就能迅速从细胞质转移到细胞核,识别并结合DNA序列激活相应基因的表达。值得注意的是,经典路径的NF-κB二聚体主要以p65/p50的形式存在,这种二聚体参与了体内几乎所有的免疫反应。
区别于经典路径,非经典路径不依赖IκB的降解,而是依赖NF-κB诱导激酶(NF-κB-inducing kinase)和IKKα的协同作用。该途径的信号来源通常来自肿瘤坏死因子家族的细胞因子,如B细胞活化因子(B-cell activating factor)和淋巴毒素-β(ymphotoxin-β),上游信号通路激活NF-κB诱导激酶,随后导致IKKα 的磷酸化,激活的NF-κB诱导激酶和IKKα促进p100的羧基末端丝氨酸残基的磷酸化,随后,p100发生泛素化,其C端的IκB样结构域发生选择性降解并生成p52,p52会与RelB结合形成非典型NF-κB复合物——p52/RelB,该复合物会在IκBδ辅助下入核,参与转录调节。
NF-κB通路与细胞焦亡
NF-κB信号通路可调节基因表达,并可影响到各种不同的生物学过程,包括先天和适应性免疫、炎症、应激反应、B 细胞发育和淋巴器官形成。同时,NF-κB与细胞焦亡有密不可分的联系,接下来我们分别介绍与NF-κB关联的关键分子及其分子机制。
NLRP3炎症小体与NF-κB
当NF-κB通路被激活,进入到细胞核后,会调节一系列基因的表达,其中的一些基因会诱导细胞焦亡的发生,比如会结合到NLRP3基因上启动子上,促进NLRP3的表达。同时,NF-κB也促进了NLRP3通路的核心成员——ASC衔接蛋白和前caspase-1的表达。NLRP3是一种细胞内受体,结构上具有独特的三聚体,具有三个核心结构域:NACHT, LRR和PYD。其中,NACHT结构域具有ATPase活性,参与NLRP3的寡聚化过程,LRR有识别信号的功能,PYD在炎症小体构建过程中参与ASC接头蛋白的PYD结构域相互作用。当细胞感知到特定的危险信号,如损伤、病原体感染或代谢不平衡时,NLRP3的PYD结构域与ASC的PYD结构域结合形成复合体颗粒(speck)。ASC接头蛋白除了具有PYD结构域外还有一个CARD结构域,NLRP3与ASC形成复合体后,CARD结构域与pro-caspase-1的CARD结构域结合(募集pro-caspase-1),最终组装成复杂的多蛋白结构,即NLRP3炎症小体(NLRP3 inflammasome )。
一旦上述的NLRP3炎症小体被激活,pro-caspase-1因在NLRP3与ASC作用下发生构象变化,pro-caspase-1发生自我剪切生成成熟体——caspase-1,caspase-1会进一步切割pro-caspase-1,触发一个正反馈循环。成熟的caspase-1会剪切GSDMD的Asp275位点,生成GSDMD-N。GSDMD-N会锚定在膜结构的磷脂上,并形成多聚体,围绕着多聚体,GSDMD-N会在细胞膜上打开一个直径10–20 nm的孔洞。孔洞会破坏细胞膜两侧的物质平衡,细胞肿胀破裂,细胞发生焦亡。同时caspase-1还能剪切并激活促炎因子pro-IL-1β 和 pro-IL-18 。当细胞因caspase-1参与下发生焦亡时,也会释放出IL-1β和IL-18,诱导临近的免疫细胞发生炎症反应。
Yuanjie Li等人研究发现1,异黄酮能通过miRNA-27a/SYK/NF-κB通路来抑制NLRP3诱导的细胞焦亡,改善患者的焦虑。ACSS2在LPS的诱导下能通过KLF5/NF-κB信号通路诱导NLRP3表达上调,促进细胞的凋亡,引起急性肾损伤2。作为研究的热门领域之一的微塑料,有报道发现微塑料能激活NF-κB-NLRP3,通路,诱导细胞焦亡和靶器官的损伤3。总而言之,NF-κB-NLRP3-caspase-1是较为常见的焦亡通路,参与众多的焦亡相关疾病的发展过程。
NF-κB 与 caspases-3/8/9
在以往的观点中认为,caspases-3/8/9是凋亡的重要调节因子。但是NF-κB能利用caspases-3/8/9介导焦亡的发生。当NF-κB进入到细胞核后,会促进pro-caspase-3 和 TNF-α的表达。激活后的caspase-3会剪切GSDMD生成GSDMD-N诱导细胞焦亡的发生。同时,上调的TNF-α能激活caspase-3 , caspase-8和 caspase-9,激活的caspase-8能剪切GSDMD和GSDMC ,并进一步激活caspase-3和caspase-9,进一步诱导焦亡的发生。此外,也有多项研究证实,NF-κB诱导上调的TNF-α也会反过来进一步激活NF-κB的通路。
在一项青光眼的研究中发现4,HMGB1响应急性眼压升高,通过NF-κB通路上调NLRP3 和caspase-8 的表达,诱导视网膜神经节细胞的焦亡。癌症治疗的,明星分子——PD-L1,近年来的研发现5,PD-L1能通过上调caspase-8 的表达诱导肿瘤细胞的焦亡。总之相当于经典的NF-κB-NLRP3-caspase-1通路,NF-κB-caspases-3/8/9通路给细胞死亡的研究开辟的新的思路和攻克疾病的的方法。
NF-κB 与caspase-4
Caspase-4是一种具有四个亚基的大分子复合物,每个亚基含有一个半胱氨酸蛋白酶活性中心。当NF-κB通路激活时,可以上调pro-caspase-4的表达,随后N端的CARD结构域能识别细胞的内的刺激信号后,会将自身转化为有活性的caspase-4,随后剪切GSDMD,诱导细胞焦亡。此外在一些压力环境下,如线粒体内膜通透性改变,也会激活caspase-4,CARD结构域能识别并结合Apaf-1形成焦亡小体(pyroptosome complex),该复合物能结合并激活caspase-3,随后,激活的caspase-3剪切GSDMD导致细胞焦亡6。
NF-κB在细胞焦亡通路中的复杂角色,如同细胞世界中的“指挥家”,精准调控着炎症与免疫的交响乐。从NLRP3炎症小体的组装到caspase家族的激活,NF-κB通过多条路径推动细胞焦亡的进程。这些发现不仅揭示了细胞死亡机制的精妙,更为疾病治疗提供了全新的靶点。汉恒生物专营病毒包装十余载,建立了庞大的基因研究现货工具库,可为广大研究人员提供组蛋白甲基化修饰相关基因的过表达病毒现货。若有其他病毒包装需求或疑问,也欢迎随时咨询汉恒生物微信公众号或拨打官网技术服务热线:400-092-0065。
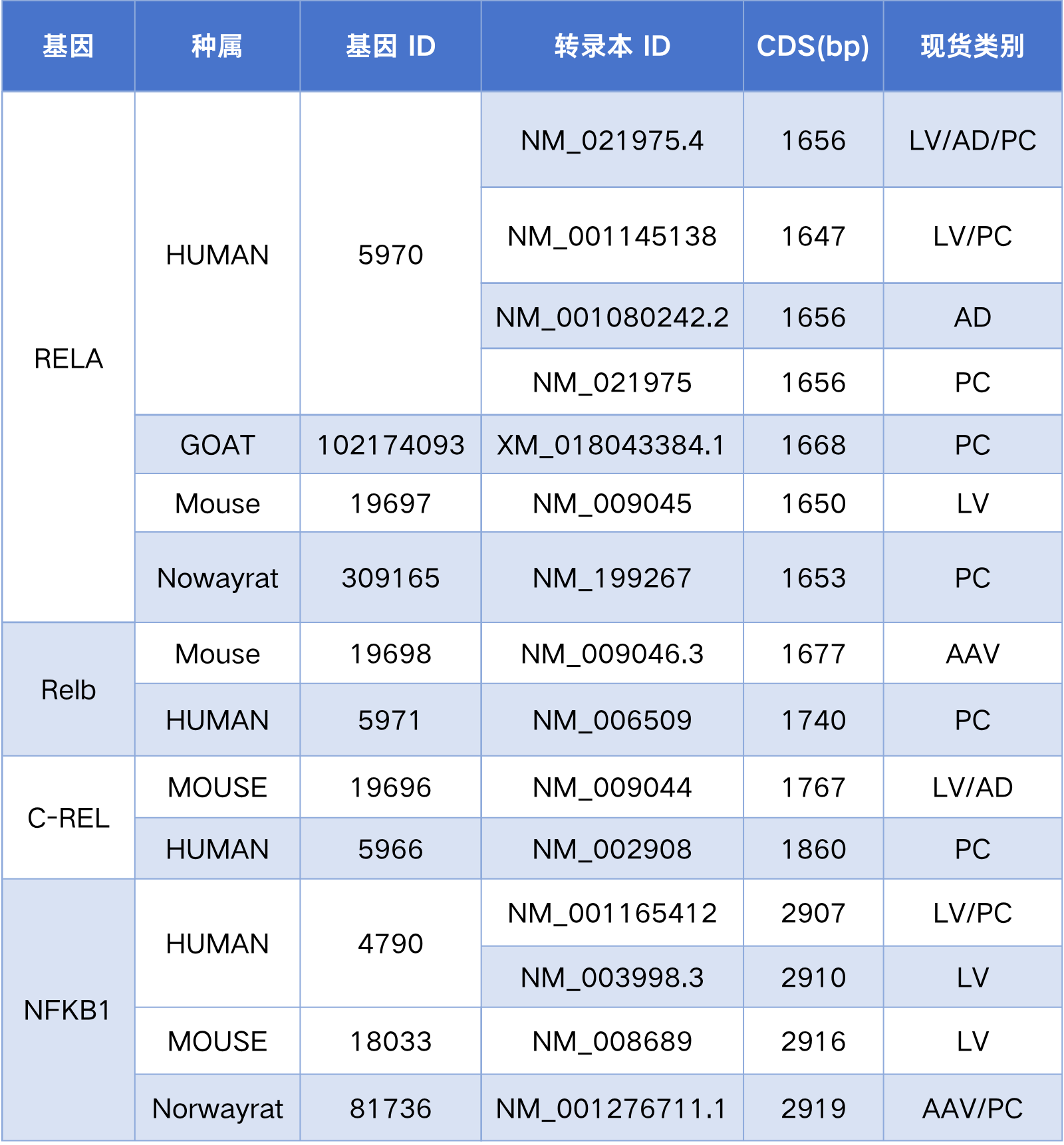
汉恒生物NF-κB相关产品现货
参考文献:
1.Li Y, Song W, Tong Y, et al. Isoliquiritin ameliorates depression by suppressing NLRP3-mediated pyroptosis via miRNA-27a/SYK/NF-κB axis. J Neuroinflammation. 2021;18(1):1.
2.Lu J, Hou Y, Liu SX, et al. Acetyl-CoA synthetase 2 induces pyroptosis and inflammation of renal epithelial tubular cells in sepsis-induced acute kidney injury by upregulating the KLF5/NF-κB pathway. Cell communication and signaling : CCS. 2024;22(1):187.
3.Zhang Y, Yin K, Wang D, et al. Polystyrene microplastics-induced cardiotoxicity in chickens via the ROS-driven NF-κB-NLRP3-GSDMD and AMPK-PGC-1α axes. Sci Total Environ. 2022;840:156727.
4.Chi W, Chen H, Li F, et al. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J Neuroinflammation. 2015;12:137.
5.Hou J, Zhao R, Xia W, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22(10):1264-1275.
6.Wei Z, Gao R, Sun Z, et al. Baicalin inhibits influenza A (H1N1)-induced pyroptosis of lung alveolar epithelial cells via caspase-3/GSDME pathway. J Med Virol. 2023;95(5):e28790.









 8325
8325










