糖尿病(Diabetes mellitus)顾名思义,尿液中含有糖分,患病者容易出现消瘦和口渴症状,我国古代又称其为“消渴症”。是一种慢性、代谢性、非传染性疾病,主要表现为“三多一少”,即多饮、多尿、多食和体重下降,当胰腺胰岛素分泌不足或机体无法有效利用胰岛素时,机体就会出现糖尿病。长期糖尿病会导致心肌细胞的结构和功能改变,而这些改变与心肌缺血或微血管粥样硬化等疾病无关,这种心肌损伤被称为糖尿病性心肌病,是糖尿病的常见并发症,据估计,超过三分之一的糖尿病病患最终都会发展为糖尿病性心肌病。心肌细胞死亡是导致糖尿病心肌损伤的核心事件,然而病患心肌细胞死亡的具体机制在很大程度上却仍然未可知。
2022年11月30日,复旦大学附属中山医院邹云增教授、葛均波院士及复旦大学基础医学院李立亮副教授合作在《Circulation》(IF=37.8)发表了题为:“Cannabinoid Receptor 2-Centric Molecular Feedback Loop Drives Necroptosis in Diabetic Heart Injuries”的研究论文,
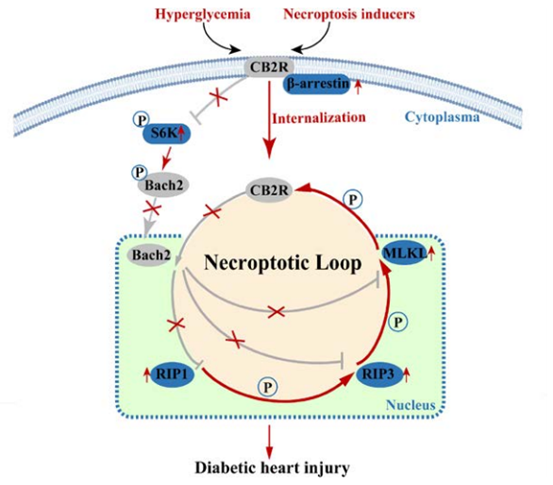
在这项研究中,作者发现在糖尿病晚期,细胞坏死性凋亡的激活与心肌损伤息息相关,大麻素受体2(CB2R)是调节坏死性凋亡的关键因子,CB2R抑制转录因子BACH2 520丝氨酸位点磷酸化,从而导致BACH2易位至细胞核并抑制细胞坏死性凋亡相关基因激酶受体互作蛋白1(RIP1)、激酶受体互作蛋白3(RIP3)和混合谱系激酶结构域样假激酶(MLKL)的表达,阐述了糖尿病通过CB2R-BACH2-MLKL引起心肌损伤的新机制,为调控糖尿病心肌损伤提供了一种有前景的方法。
具体研究内容,就让小编带大家一起了解一下吧!
提示:本研究中使用的9型腺相关病毒(AAV9)由汉恒生物提供技术支持,详情可咨询https://www.hanbio.net/小恒为您提供专业服务(⑅˃◡˂⑅)
首先作者通过查阅资料发现糖尿病导致的心肌损伤过程中细胞凋亡和坏死性凋亡是重要原因,因此作者对HG处理的心肌细胞的细胞凋亡和坏死性凋亡水平进行检测,发现无论是原代还是体外培养的小鼠心肌细胞系(HL-1)经HG处理后凋亡和坏死性凋亡均可被激活,12-72h内细胞凋亡逐渐增强,而72h后凋亡逐渐减弱,磷酸化的RIP1、RIP3和MLKL表达量升高,TNF受体相关的死亡结构域蛋白(TRADD)和死亡域蛋白(FADD)表达量升高,此时坏死性凋亡被激活。扫描电镜下可见HG刺激72h后细胞破裂,发生坏死性凋亡,Western Blot结果也显示坏死性凋亡普遍发生在16周龄以上的糖尿病小鼠中。除此之外流式分析、免疫荧光和活细胞实时监测结果也表明,细胞坏死性凋亡主要发生在糖尿病后期。
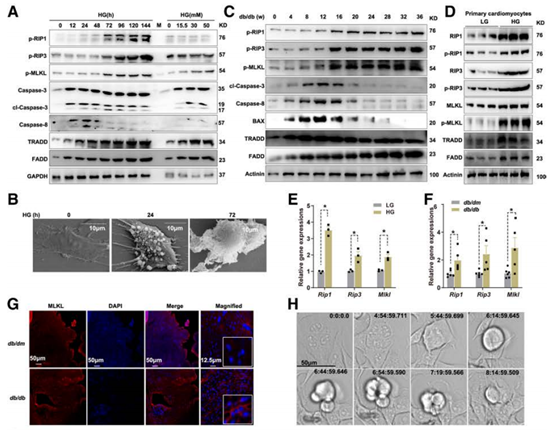
图1 糖尿病心肌损伤后期坏死性凋亡被激活
Nec-1是一种坏死性凋亡抑制剂,为了探究Nec-1抑制坏死性凋亡的治疗意义,作者对16周龄db/db小鼠注射Nec-1,超声心动图显示,尽管糖尿病小鼠与对照组相比,心功能显著受损,但Nec-1注射后心脏受损程度却得到减轻,心脏重量与胫骨长度的比值降低,FN、白介素6(IL-6)和磷酸化的RIP1、RIP3和MLKL等基因表达水平下降,坏死性凋亡信号通路被阻断,此种现象证明了在坏死性凋亡发展阶段对糖尿病进行干预可以为心脏提供有效保护。
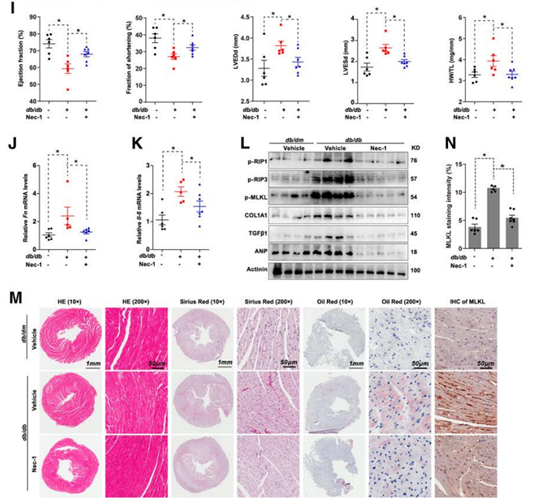
图2 Nec-1抑制糖尿病心肌损伤
为探究HG是如何激活坏死性凋亡的,作者使用G蛋白偶联受体(GPCR)激动剂添加到HG处理的HL-1细胞中,结果显示GPCR中CB2R的激动剂对细胞坏死性凋亡有很好的抑制作用,两种CB2R激动剂AM1241和JWH-133均能抑制HG诱导的细胞坏死性凋亡,其中JWH-133对细胞坏死性凋亡抑制效果在CB2R过表达后进一步增强,在CB2R敲低后抑制作用减弱。在HL-1细胞中也发现类似的现象。然后,作者将CB2R激动剂用于糖尿病小鼠,Western Blot、组织学研究、超声心动图结果均显示,CB2R激动剂改善了糖尿病小鼠心肌损伤,且伴随着RIP1、RIP3和MLKL的总蛋白以及磷酸化的蛋白量下降。免疫组化结果显示,在糖尿病小鼠心脏中MLKL表达上调并发生磷酸化,CB2R激动剂可逆转此现象。扫描电镜观察进一步证实,CB2R激动剂AM1241可恢复HG导致的细胞成团现象。这些结果均表明,CB2R激动剂可有效抑制糖尿病小鼠心肌细胞坏死性凋亡。
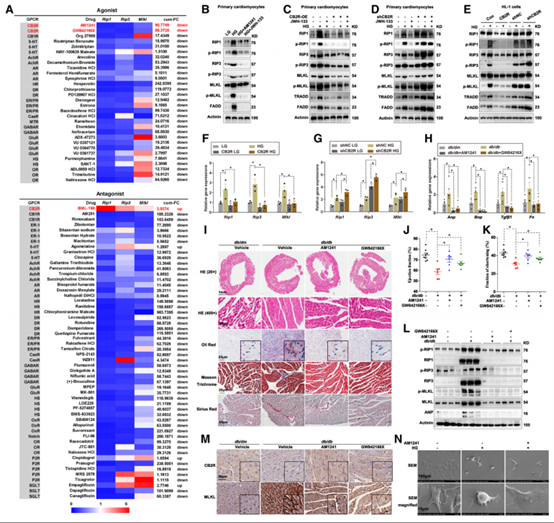
图3 CB2R有效抑制糖尿病心肌坏死性凋亡
通过RNA-seq作者筛选了可能参与其过程的转录因子,其中BACH2沉默后逆转了CB2R对细胞坏死性凋亡的抑制作用,生物信息学分析发现BACH2上存在2个与RIP1和RIP3启动子结合的位点,进一步进行染色质免疫共沉淀实验、定点突变实验和双荧光素酶实验结果表明,BACH2可与RIP1和RIP3启动子的特定区域结合抑制靶基因表达,从而抑制细胞坏死性凋亡,后对BACH2进行定位分析,发现BACH2上游S6激酶(S6K)磷酸化后阻止了BACH2细胞质向细胞核中转运,而CB2R抑制S6K蛋白的磷酸化促进BACH2的胞浆转位,维持BACH2发挥功能,从转录水平抑制坏死性凋亡通路的激活。
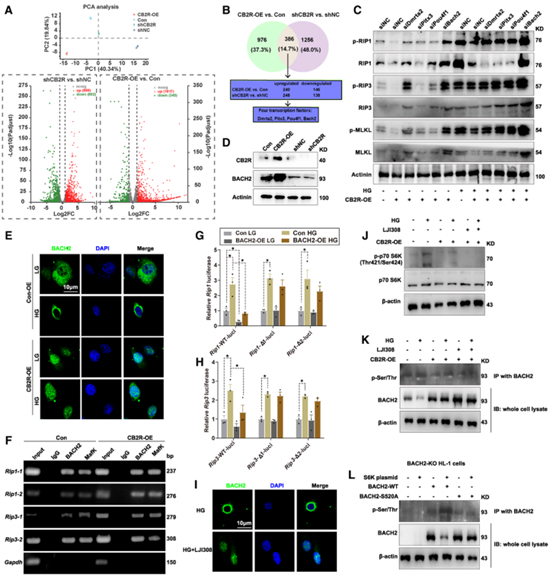
图4 CB2R调节BACH2抑制坏死性凋亡
在体外培养的细胞系中,作者观察到敲低CB2R通常会促进HL-1坏死性凋亡水平,但是当BACH2过表达后此种现象被逆转。因此作者验证在糖尿病小鼠是否也存在此现象,糖尿病小鼠心脏原位注射AAV9-α-MHC–BACH2以过表达BACH2,观察到CB2R拮抗剂AM630可加重糖尿病小鼠心肌损伤,但是BACH2过表达小鼠中心脏损伤效果明显减轻。WB检测发现使用AM630后FN、IL-6、RIP1、RIP3和MLKL表达上调,BACH2过表达后上调被抑制。免疫荧光染色、组织学检查结果也表明,AM630加重了糖尿病引起的炎症和纤维化,而BACH2可有效缓解心脏损伤。这些现象均表明,抑制CB2R则加重糖尿病心脏损伤,BACH2过表达可以逆转此种现象。
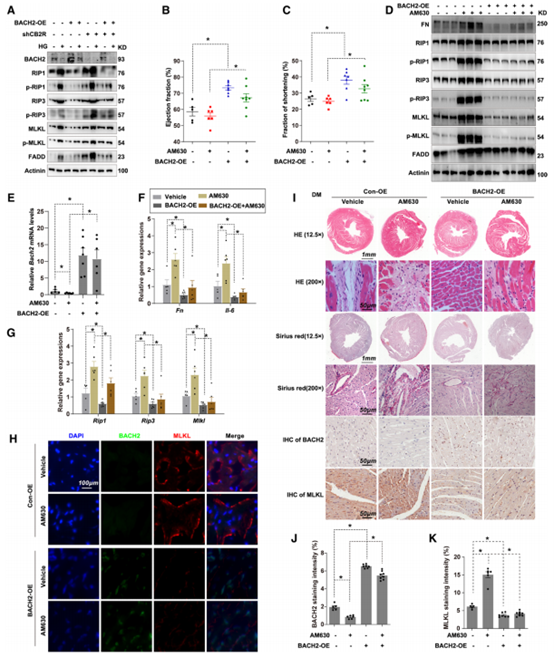
图5 过表达BACH2抑制CB2R拮抗剂诱导的糖尿病心脏损伤
为进一步探究CB2R调控BACH2的作用机制,作者构建了BACH2敲除的糖尿病小鼠(BACH2-CKO),其FADD、IL-6、α1-1型胶原基因(COL1A1)、RIP1、RIP3和MLKL的总蛋白和磷酸化的蛋白表达量升高。值得注意的是,AM1241抑制细胞坏死性凋亡并导致IL-6和COL1A1表达量降低的现象仅发生在糖尿病小鼠中,而在BACH2-CKO糖尿病小鼠中则未出现此现象,超声心动图和组织学研究结果也显示AM1241仅能改善糖尿病小鼠心肌损伤。这些现象表明CB2R改善糖尿病小鼠心肌损伤过程中需要BACH2的存在。
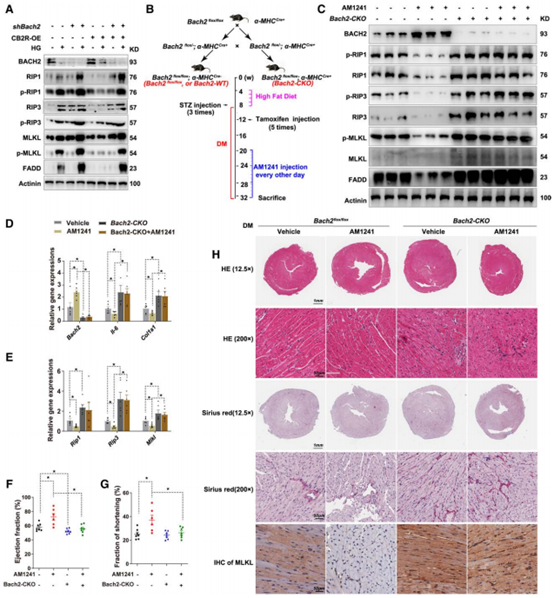
图6 BACH2敲除加重糖尿病心肌损伤并抑制CB2R对心脏的保护作用
体内和体外实验的结果表明HG环境下细胞膜上的CB2R显著降低,而细胞质中CB2R增加,CB2R的含量和活性在很大程度上取决于其亚细胞定位和磷酸化修饰,而CB2R又与绝大多数GPCR一样可以结合β-arrestin 2,因此作者检测了β-arrostin 2的表达量,发现早在HG处理后12小时β-arrestin 2开始增加,CB2R开始降低。MLKL是CB2R丝氨酸352(S352)的上游激酶,作者自制了特异性靶向CB2R(S352)位点的磷酸化抗体以检测p-CB2R表达水平。结果表明,HG可增加p-CB2R表达水平,降低总CB2R表达水平,MLKL、RIP1或RIP3过表达都可促进HEK-293T细胞中CB2R(S352)位点的磷酸化,而MLKL磷酸化后,会伴随CB2R泛素化增强。这些结果表明,MLKL在S352位点使CB2R磷酸化并导致CB2R降解。
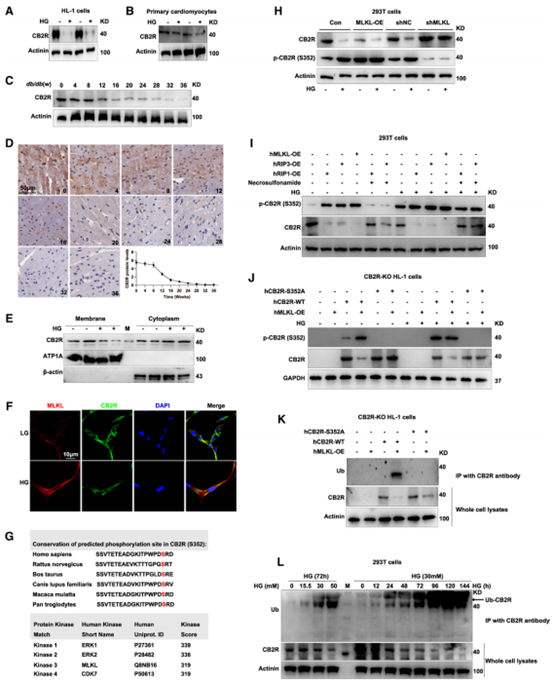
图7 CB2R与β-arrestin 2结合在S352被磷酸化并在HG环境下被MLKL降解
收集已故有无糖尿病史的人群心脏样本,作者分析发现糖尿病心脏样本中,CB2R和BACH2显著降低,而TRADD和FADD显著增加,心脏伴有明显的纤维化。免疫组化结果显示,糖尿病心脏样本中CB2R和BACH2蛋白水平显著降低,而MLKL蛋白水平显著升高。这些发现表明,糖尿病与这些蛋白质的表达有关。免疫荧光结果显示人类糖尿病心脏中RIP1、RIP3和MLKL表达量升高,CB2R或BACH2与MLKL表达水平以及心脏纤维化程度呈显著负相关。
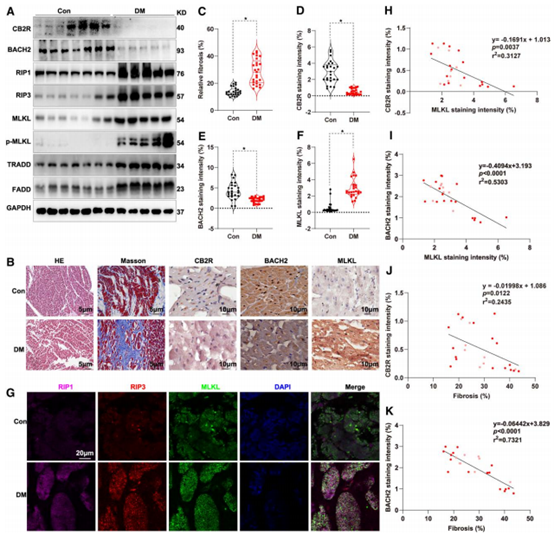
图8 CB2R和BACH2与糖尿病心肌损伤呈负相关
综上所述,作者证明了坏死性凋亡发生在糖尿病心肌损伤晚期,CB2R可通过BACH2抑制坏死性凋亡进而缓解糖尿病心肌损伤的进展,而坏死性凋亡通路的效应分子MLKL可促进CB2R(S352)位点磷酸化最终导致其降解,这些结果表明CB2R-BACH2-MLKL形成了一个坏死性凋亡新型环路,为开发新的糖尿病心肌损伤干预靶点提供理论支撑。









 8325
8325










